



分享:MgO改性HA對Mg-Zn-Zr/m-HA復合材料組織及性能的影響

鄭浩然
摘要
為改善復合材料中納米增強體易團聚的問題,將陶瓷納米棒HA進行表面包覆MgO改性處理(m-HA),并采用高熔體剪切攪拌技術制備Mg-3Zn-0.8Zr合金(MZZ)、Mg-3Zn-0.8Zr/1HA復合材料(MZZH)和Mg-3Zn-0.8Zr/1m-HA復合材料(MZZMH)。研究了m-HA對Mg-Zn-Zr/HA復合材料微觀組織、力學性能和耐蝕性能的影響。結果表明,陶瓷納米棒HA的加入細化了MZZ合金的組織,提高了MZZ合金的力學性能和電化學耐蝕性能。與MZZH相比,MZZMH的晶粒更加細小均勻,陶瓷納米棒在基體中的分布更均勻。擠壓態MZZMH的力學性能較MZZH顯著提高,其硬度、屈服強度、抗拉強度和延伸率分別達到92 HV、291 MPa、325 MPa和8.62%。MZZMH的自腐蝕電位比MZZH高59 mV,MZZMH的腐蝕速率較MZZH降低,在SBF中浸泡7 d后穩定在5 mm/a。腐蝕機理的不同使MZZMH復合材料的耐蝕性能優于MZZH。因此,MgO改性可有效促進HA納米棒的均勻分布,進而顯著提高MZZMH的力學性能和耐蝕性。
關鍵詞:
Mg及其合金因其良好的生物可降解性及與人體骨相匹配的力學性能,作為短期行使功能的植入材料應用前景廣闊[1-3]。但其強度偏低、塑性不足、腐蝕速率過快的問題仍需改善,以促進生物醫用鎂合金的臨床應用進展[4]。近年來,鎂基復合材料因其可調可控的力學性能和耐蝕性能逐漸成為該領域研究的熱點之一[5-8]。復合材料中陶瓷顆粒增強體不僅在鎂合金基體中阻礙位錯運動,提高屈服強度,并可作為異質晶核,降低形核功,增大形核率,起到細化晶粒作用。而細晶結構鎂基復合材料在體內外更趨向均勻腐蝕,進而提高鎂基體的耐蝕性[9]。增強體的種類、含量、尺寸及分散性是影響鎂基復合材料性能的重要因素。Zhao等[10]制備了CNTs/AZ91D復合材料,碳納米管(CNT)的加入提高了復合材料的力學性能,但由于CNT與基體無化學反應,界面結合狀態不甚理想。一些研究者以羥基磷灰石(HA)和原位生成的MgO為增強體,用粉末冶金法制備鎂基復合材料,其抗拉強度、硬度和耐蝕性均隨增強體含量的增加而提高,但塑性顯著降低,綜合性能不夠理想[11,12]。原因是復合材料中所涉及的納米增強體均出現了較嚴重的顆粒團聚現象,且與基體之間界面非冶金態結合。由于生物活性陶瓷顆粒與Mg熔體的浸潤性差,難以通過機械攪拌混合均勻,并直接影響材料性能,因此,解決納米陶瓷顆粒在鎂基復合材料中的團聚問題迫在眉睫[13]。研究者嘗試采用增強體顆粒表面包覆或改變復合材料制備技術克服團聚。Gu等[14]用熔體滲透的方法將Mg-Ca合金滲透到多孔支架HA/磷酸鈣(TCP)中制備復合材料,其耐蝕性比Mg-Ca合金提高了68%,但力學性能卻下降了50%。Ye等[15]用明膠對納米HA進行表面改性,采用真空與惰性氣體保護方法制備了1%HA/Mg-Zn-Zr復合材料,強度和塑性同步提高,但局部團聚現象依然存在。Liu等[16]采用熔體強力剪切攪拌技術,利用剪切應變破碎團聚顆粒,顯著提高了β-TCP顆粒在鎂基復合材料中的分散性。
為進一步提高納米增強體的浸潤性而又不引入其它雜質,采用MgO對納米增強體進行表面包覆改性應是一個有效的方法。這是由于MgO是一種相對重量小、與基體變形協調性好的生物可降解材料。此外,作為Mg基體的同源材料,MgO與鎂基體間有著良好的晶體取向關系,即半共格界面。因此在鎂合金熔煉過程中,MgO可極大增加陶瓷納米棒與Mg液的浸潤度,從而實現納米棒HA的有效分散,解決團聚的問題。
在本課題組以往工作[17-19]的基礎上,本工作以MgO改性前后的HA納米棒為增強體,Mg-3Zn-0.8Zr為基體,采用高熔體剪切攪拌技術熔煉復合材料,對比研究MgO改性對Mg-Zn-Zr/HA復合材料微觀組織、力學性能及耐蝕性能的影響,以求獲得有效改善納米陶瓷增強體在鎂基體中分散不均的工藝方法。
采用水熱法及化學反應相結合的工藝制備MgO改性HA納米棒[20]。在攪拌條件下,將濃度為0.67 mol/L的C6H5Na3O72H2O溶液10 mL逐滴加到濃度為0.67 mol/L的15 mL Ca(NO3)2溶液中,充分攪拌,逐滴加入濃度為0.4 mol/L的Na3PO4溶液15 mL,繼續攪拌后,將混合液轉移至50 mL的反應釜內襯中,于150 ℃反應24 h。取出反應液,將濃度為1 mol/L的Mg(NO3)26H2O溶液8 mL逐滴加入上述混合溶液,經過靜置、離心、干燥及300 ℃煅燒,得到MgO包覆HA納米棒,計作m-HA。
以工業高純Mg (99.99%)、高純Zn (99.99%)、Mg-Zr中間合金(30%Zr,質量分數)、HA納米棒和m-HA納米棒為原料,采用0.4%SF6+99.6%N2混合氣氛保護,分別熔煉Mg-Zn-Zr合金及其復合材料。將純Zn和Mg-Zr中間合金按設計比例加入Mg熔體中保持15 min使其熔化并充分擴散。HA和m-HA納米棒于200 ℃預熱2 h,在670 ℃時分別加入Mg-Zn-Zr合金熔體中,熔體升溫至720 ℃,用英國布魯奈爾大學[21]先進凝固技術研究中心(BCAST)開發的高熔體剪切攪拌設備充分攪拌5~10 min,然后澆鑄成直徑60 mm的鑄錠,分別記為Mg-3Zn-0.8Zr (MZZ)合金、Mg-3Zn-0.8Zr/1HA (MZZH)及Mg-3Zn-0.8Zr/1m-HA (MZZMH)復合材料。3種鑄錠在350 ℃下預熱2 h后熱擠壓(YQ 32-315)成直徑8 mm的棒材,擠壓比為1:56。
通過U-TV0.5VC-3金相顯微鏡(OM)、JSM-6700F掃描電子顯微鏡(SEM)及Tencnai G2 F20透射電子顯微鏡(TEM)觀察材料的顯微組織。采用SEM及TEM裝配的X射線能譜儀(EDS)分析各相化學成分。采用D/max 2500 PC X射線衍射分析儀(XRD)測量試樣的物相,CuKα,波長0.15418 nm,測試速率5°/min,并用Jade 6.0軟件分析衍射圖譜和相。
采用HMV-2T Vickers硬度計測量材料硬度,設置載荷9.8 N,保持時間20 s,取每個樣品中6個不同部位硬度的平均值。根據GB/T16865-1997加工標準拉伸試樣,用WDW-100電子萬能試驗機進行拉伸實驗,拉伸速率為0.5 mm/min,每種材料以3個平行試樣的平均值作為實驗結果。
合金及復合材料冷卻曲線的測量由自制裝置完成,用13 mm厚的隔熱層將鋼制坩堝放入電阻爐中預熱至700 ℃,將完全熔化的MZZ或MZZH熔體倒入坩堝中。為防止冷卻速率過快,將鋼制坩堝側面和底部圍上隔熱材料并置于隔熱磚上,在氣氛保護下,將與電腦連接的0.5 mm熱電偶插入坩堝的正中間,利用軟件記錄熔體溫度隨時間變化的曲線。
為測試材料的電化學性能,將擠壓態MZZ、MZZH和MZZMH機加工成直徑8 mm、厚3 mm的圓片,并用3000號砂紙打磨試樣表面至光亮。實驗在模擬體液(SBF)中進行,SBF (pH=7.4,37 ℃)由3.273 g/L NaCl、1.134 g/L NaHCO3、0.186 g/L KCl、0.134 g/L Na2HPO47H2O、0.152 g/L MgCl26H2O、0.184 g/L CaCl22H2O、0.036 g/L Na2SO4和3.029 g/L (CH2OH)5CNH2組成。將圓片和導電性好的Cu導線焊接,用Zahner Zennium電化學工作站的標準三電極體系測量樣品的極化曲線和電化學阻抗譜(EIS),試樣作為工作電極,石墨電極作為輔助電極,飽和甘汞電極(SCE)作為參比電極,掃描速率1 mV/S。測試前試樣在SBF中浸泡30 min以獲得穩定的開路電位(OCP),以3個平行試樣的平均值作為實驗結果。
體外浸泡實驗在SBF中進行,將直徑8 mm、厚3 mm的MZZMH及MZZH圓片浸泡在40 mL的SBF中,37 ℃恒溫水浴震蕩器中震蕩,2 d更換1次SBF。前36 h內每隔2 h用STARTER 3100 pH劑(OHOUS)測定試樣pH值,浸泡3、5和7 d后再測定1次,計算3個平行試樣的均值。浸泡前期每隔2 h用自制的析氫裝置測定浸泡反應產生的H2量,浸泡3、5和7 d后再測定1次,最終值取3個平行試樣的平均值。根據測定的析氫量換算出試樣的質量損失,根據式(1)計算腐蝕速率v (mm/a):
式中,?W為試樣浸泡前后的質量損失(g),A為試樣與SBF的接觸面積(mm2),t為浸泡時間(a),D為Mg的標準密度。將MZZMH及MZZH試樣浸泡6 h、12 h、1 d、3 d、5 d及7 d,取出后干燥,采用體式顯微鏡和SEM觀測浸泡試樣的表面形貌,并結合EDS分析試樣表面沉積物的成分。用鉻酸洗液去除浸泡試樣表面的沉積物,采用SEM觀測其表面形貌。去腐蝕產物的溶液由5 g重鉻酸鉀溶液、10 mL蒸餾水和90 mL濃硫酸配制而成。每個浸泡時間準備3個平行試樣,取平均值以確保實驗結果準確性。
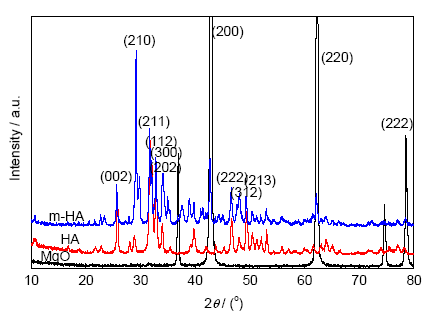
圖1是MgO、HA及m-HA的XRD譜。參照JCPDS卡片,m-HA的譜圖中2θ角對應衍射峰的位置與標準HA的主衍射峰一致,但峰位整體輕微左移,且譜圖中3個衍射峰的位置分別對應標準MgO衍射峰3個主要晶面衍射,其峰位整體略有右移,這說明MgO不僅僅是機械包裹在HA表面,而是兩者在界面產生一定的離子交換,確保了二者之間的界面結合。由圖1中還可看出,HA改性后,(210)晶面的衍射峰強度增加,說明MgO改性HA后晶體易沿(210)晶面生長,增加m-HA納米棒的長度。
圖1 MgO、HA、m-HA的XRD譜
Fig.1 XRD spectra of MgO, HA and modified HA (m-HA)
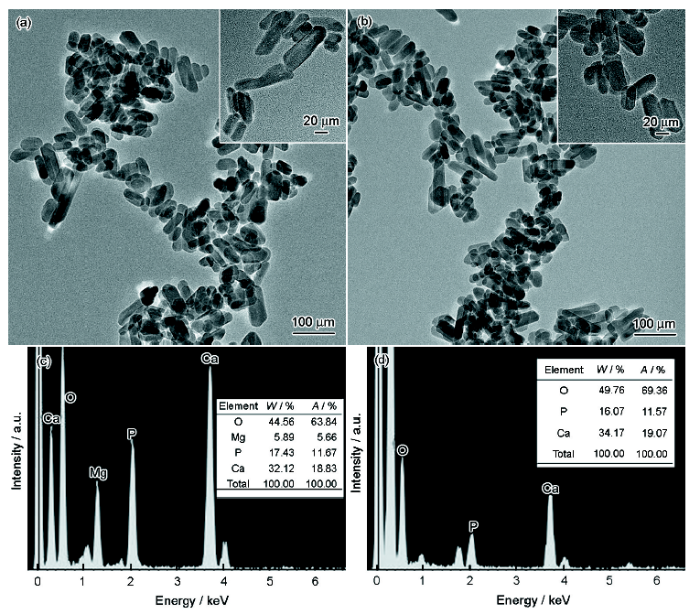
圖2是HA包覆MgO改性前后的TEM像及其EDS譜。從圖2a和b可以看出,m-HA納米顆粒的分散性略微好于HA納米顆粒,二者均為棒狀顆粒,m-HA的平均長度約為53 nm,HA為37 nm。對納米HA及m-HA顆粒進行EDS分析,從圖2c和d可以得出,HA中主要含有O、P、Ca元素,m-HA中主要含有O、Mg、P、Ca元素。結合XRD譜推斷制備的m-HA納米顆粒確實在HA的外面包覆上了MgO。
圖2 m-HA和HA納米顆粒的TEM像及EDS譜
Fig.2 TEM images (a, b) and EDS (c, d) of m-HA (a, c) and HA (b, d) nanoparticles (Insets in Figs.2a and b are enlarged images, W—mass fraction, A—atomic fraction)
鑄態MZZMH、MZZH復合材料和MZZ合金的OM像如圖3所示。3種材料的鑄態組織主要由α-Mg等軸晶組成,其平均晶粒尺寸依次為14.4、20.6和62.3 μm。顯然,復合材料的晶粒尺寸遠小于MZZ合金,說明HA納米顆粒細化晶粒效果顯著。與MZZH相比,MZZMH的晶粒尺寸細小、分布比較均勻、陶瓷顆粒和第二相均勻地分布在晶粒內和晶界上。MZZH中顆粒的團聚現象更明顯,這說明僅靠熔煉時的機械攪拌來打碎團聚的HA是遠遠不夠的。
圖3 鑄態MZZMH、MZZH和MZZ的微觀組織OM像
Fig.3 OM images of as-cast Mg-3Zn-0.8Zr/1m-HA (MZZMH) (a), Mg-3Zn-0.8Zr/1HA (MZZH) (b) and Mg-3Zn-0.8Zr (MZZ) (c)
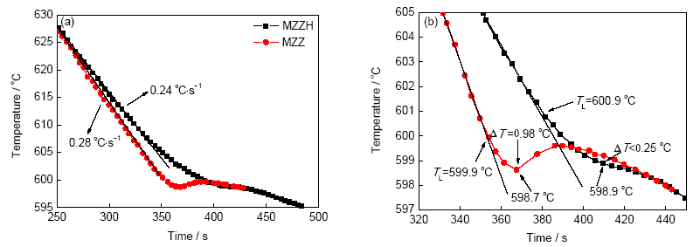
圖4為MZZ和MZZH試樣的冷卻曲線。基于圖4a,MZZ合金的冷卻速率約為0.28 ℃/s,與MZZH復合材料的冷卻速率(0.24 ℃/s)極為相似。圖4b為靠近液相線的測量數據。MZZ合金的過冷度?T約為0.98 ℃,但加入1%HA后的過冷度卻大幅減小(?T<0.25 ℃)。這表明,在鎂合金熔體中,HA顆粒的存在可以加強非均勻形核過程的進行,減小鑄件的晶粒尺寸,這也與本工作中測量的晶粒尺寸結果一致。
圖4 MZZ和MZZH的冷卻曲線
Fig.4 Cooling curves for MZZ and MZZH (a) and corresponding enlarged curves (b) (?T—degree of under cooling, TL—actual solidification temperature)
擠壓態復合材料MZZMH和MZZH的OM及SEM像如圖5所示。MZZMH和MZZH在擠壓后均發生了完全動態再結晶,且陶瓷顆粒及第二相在擠壓過程中被破碎并沿擠壓方向呈帶狀分布。MZZMH和MZZH的平均晶粒尺寸分別為2.4和3.2 μm。與MZZH相比,在MZZMH中陶瓷顆粒沿晶界分布更均勻,沿擠壓流線方向呈條塊狀不連續分布的陶瓷顆粒及第二相的尺寸更小、分布更均勻,更容易通過陶瓷顆粒及第二相激發形核機制(PSN)促進動態再結晶的發生而進一步細化晶粒[22]。這說明納米陶瓷顆粒HA的改性會使MZZH的晶粒得到細化,組織分布更加均勻。
圖5 擠壓態MZZMH和MZZH復合材料的OM和SEM像
Fig.5 OM (a, d) and SEM images (b, c, e, f) of as-extruded MZZMH (a~c) and MZZH (d~f)
2.3.1 MZZ、MZZH及MZZMH的力學性能
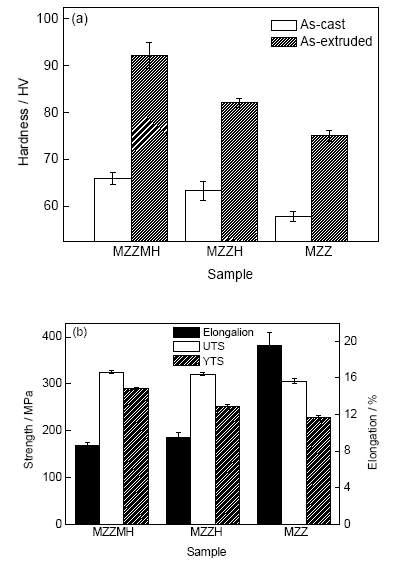
圖6a為鑄態及擠壓態MZZMH、MZZH及MZZ的Vickers硬度。可以看出,鑄態MZZMH (65.82 HV)和MZZH (63.28 HV)的Vickers硬度基本相當,但均高于MZZ (57.82 HV)。經過熱擠壓后復合材料的Vickers硬度有明顯提高,在擠壓過程中,復合材料發生塑性變形,得到更加細小均勻的等軸晶,細化了晶粒,增加了晶界密度,提高了材料的硬度。擠壓態MZZMH的Vickers硬度明顯高于MZZH,分別達到92.23和82.05 HV。擠壓態復合材料的Vickers硬度均遠遠大于MZZ合金(75 HV),說明HA及m-HA對Mg-Zn-Zr合金的硬度有積極影響。
圖6 MZZMH和MZZH復合材料的力學性能
Fig.6 Hardness (a) and tensile properties (b) of MZZMH和MZZH composites (UTS—ultimate tensile strength, YTS—yield tensile strength)
擠壓態MZZMH、MZZH及MZZ沿擠壓方向拉伸的性能如圖6b所示。由圖可知,材料的拉伸強度MZZMH>MZZH>MZZ,其中MZZMH的屈服強度和抗拉強度分別為291和325 MPa,分別比MZZH提高40和5 MPa,而MZZ的屈服強度(228 MPa)和抗拉強度(306 MPa)最低。MZZMH和MZZH的伸長率分別為8.62%和9.48%,兩者相差不大。綜上,HA的加入提高了MZZH的強度,HA的表面包覆上MgO后,復合材料MZZMH屈服強度的提高尤為顯著。
2.3.2 MZZ、MZZH及MZZMH的電化學測試
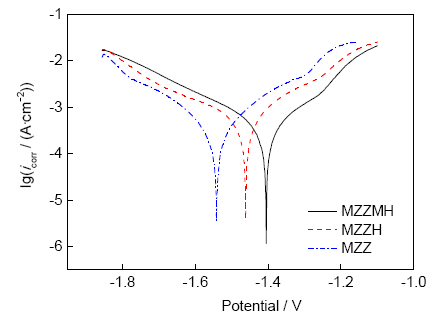
圖7給出了擠壓態MZZMH、MZZH及MZZ在SBF中的極化曲線。由于鎂合金的腐蝕存在“負差異效應”,雖然依照傳統極化曲線方法推算鎂合金的腐蝕速率并不可靠,但仍然可以用其判斷該合金的腐蝕傾向[23]。從圖7可以看出,3種材料的陽極極化曲線均有波動,說明試樣發生了點蝕。一般陽極極化曲線代表Mg2+的溶出,陰極極化曲線代表析氫反應[24],可以看出,自腐蝕電位(Ecorr)相同時,MZZH的腐蝕電流密度(icorr)比MZZMH高,比MZZ低。由極化曲線計算得到的Ecorr、陰極Tafel坡(βc)、陽極Tafel坡(βa)、極化電阻(Rp)和相應的腐蝕率(Pi)數據如表1所示。其中Rp和Pi (與icorr (mA/cm2)有關)根據以下方程[25,26]計算得到:
經過分析得到MZZMH、MZZH及MZZ的Ecorr分別為-1403、-1462和-1542 mV。一般Ecorr越低,材料的腐蝕越嚴重越迅速。材料的極化電阻Rp滿足:MZZMH>MZZH>MZZ。即HA的添加降低了MZZH的腐蝕速率,HA的改性增加了復合材料MZZMH的鈍化能力,提高了耐蝕性。
圖7 擠壓態MZZMH、MZZH及MZZ的極化曲線
Fig.7 Tafel polarization curves of as-extruded MZZMH, MZZH composites and MZZ alloy (icorr—corrosion current)
表1 擠壓態MZZMH、MZZH及MZZ在模擬體液(SBF)中的電化學參數
Table 1 Electrochemical parameters of MZZMH, MZZH and MZZ in simulated body fluid (SBF) acquired from the polarization test and EIS
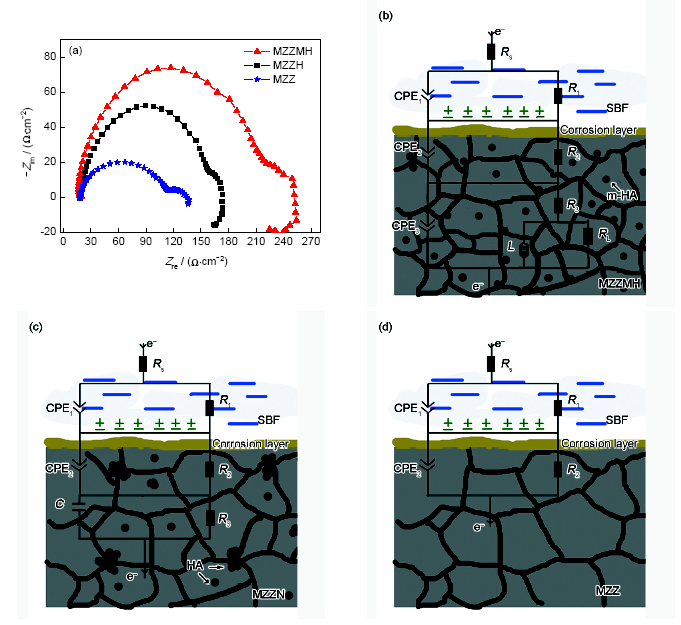
擠壓態MZZMH、MZZH及MZZ在SBF中的EIS及其擬合所用等效電路如圖8所示。通常,交流阻抗曲線由2個電容弧(位于高頻區和中頻區)和1個電感弧(位于低頻區)組成。高頻區的容抗弧表征的是由電荷傳遞電阻和雙電層電容構成的阻容弛豫過程,高頻區的容抗弧表征的是Mg腐蝕中間產物產生的容抗。電容弧和電感弧直徑的大小直接反應材料的耐腐蝕性,直徑越大,材料的腐蝕速率就越低[27]。從圖8可以看出,MZZMH的電容弧直徑大于MZZH,說明相同條件下MZZMH腐蝕過程中電荷轉移相對較少,具有較好的耐蝕性能。3種材料結合EIS的電化學反應機理(圖8b~d)表明,材料的腐蝕區別主要在于添加的納米陶瓷顆粒。圖中,Rs表示溶液的電阻;R1表示電荷轉移電阻;R2表示腐蝕產物層電阻;R3表示物質傳輸電阻,Q為角相位元件,表示一種等效元件CPE,C為常相位角元件。表1給出了擠壓態MZZMH、MZZH及MZZ在SBF中的電化學參數。對比表中數據發現,MZZ的R1、R2、R3值最小,即耐蝕性最差。MZZMH的R1、R2、R3值高于MZZH,這說明和MZZH相比,MZZMH中電荷轉移的阻力大,試樣表面形成的腐蝕層致密,表面的腐蝕比較均勻、點蝕少。所以HA的改性提高了復合材料的耐蝕性能。
圖8 擠壓態MZZMH、MZZH及MZZ的交流阻抗譜及其擬合所用等效電路
Fig.8 EIS (a) and equivalent circuit (b~d) of as-extruded MZZMH (b), MZZH (c) composites and MZZ (d) alloy (Rs—solution resistance, R1—charge transfer resistance, R2—corrosion product layer resistance, R3—mass transfer resistance, RL—corresponding resistance of inductance components, CPE—equivalence element, C—capacitance, L—inductance element)
圖9a1~a4和b1~b4分別為擠壓態MZZMH和MZZH在SBF中浸泡1、3、5和7 d后的體式顯微鏡照片。隨著材料在SBF中浸泡時間的延長,試樣表面腐蝕程度不斷加強,并不斷產生點蝕導致基體剝落。但在相同條件下,MZZH試樣表面的腐蝕坑比MZZMH更深更大,浸泡后MZZMH試樣的剩余量明顯多于MZZH。說明MZZMH在SBF中的腐蝕比MZZH緩慢。圖9c1~c4和d1~d4分別為擠壓態MZZMH和MZZH在SBF中浸泡并去腐蝕產物后形貌的SEM像。觀察發現,樣品表面存在點蝕坑,說明材料在SBF中會發生點蝕。腐蝕首先沿著晶界發生,說明材料在SBF中發生了電偶腐蝕。對比2種復合材料,當試樣在SBF中浸泡1 d時,試樣表面的腐蝕坑均勻而微小,但MZZH的腐蝕坑更寬更深。對于同種材料,隨著浸泡時間的延長,試樣表面逐漸凹凸不平,腐蝕坑尺寸加大。這說明MZZMH的腐蝕更均勻、耐蝕性更好。
圖9 擠壓態MZZMH和MZZH在SBF中浸泡不同時間的體式顯微鏡照片及去腐蝕產物后的SEM像
Fig.9 OM images (a1~a4, b1~b4) and SEM images (c1~c4, d1~d4) of surface morphologies in MZZMH (a1~a4, c1~c4) and MZZH (b1~b4, d1~d4) before (a1~a4, b1~b4) and after (c1~c4, d1~d4) removing the corrosion products for immersion 1 d (a1~d1), 3 d (a2~d2), 5 d (a3~d3), 7 d (a4~d4) in SBF
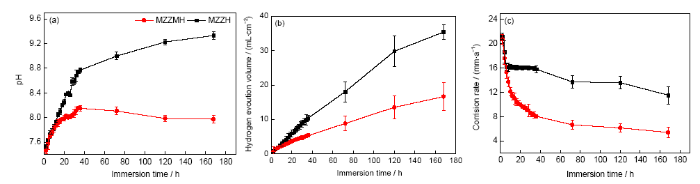
擠壓態MZZMH和MZZH在SBF中浸泡不同時間的pH值和析氫量的變化分別如圖10a和b所示。3種曲線都呈先增加后趨于平衡的趨勢。材料浸泡在SBF中會發生如下反應:
隨浸泡時間的增加,溶液中的pH值及析氫量均呈上升趨勢,腐蝕產物層有保護膜的作用,其厚度的增加會減緩材料的腐蝕。但MZZMH的pH值和析氫量一直低于MZZH,浸泡7 d后MZZMH和MZZH的pH值分別趨于8和9.3,析氫量分別穩定于16.6和35.4 mL/cm2。圖10c給出了擠壓態MZZMH和MZZH在SBF中浸泡不同時間的腐蝕速率曲線。可以看出,2種試樣的腐蝕速率曲線均為前期降低較快,后期趨于平衡,MZZMH的腐蝕速率一直低于MZZH,最后MZZMH和MZZH的腐蝕速率分別穩定在5和12 mm/a。比較可知,MZZMH的耐蝕性優于MZZH,這也與電化學結果相一致。
圖10 擠壓態MZZMH和MZZH在SBF中的pH值、析氫及腐蝕速率曲線
Fig.10 Curves of pH (a), hydrogen evolution (b) and corrosion rate (c) of as-extruded MZZMH and MZZH after immersed in SBF
對2種材料在SBF中浸泡的腐蝕表面形貌進行SEM觀察,并對其表面腐蝕產物進行EDS分析,以分析MZZH和MZZMH耐蝕性差異的原因。由圖11a~e可知,MZZH在SBF中浸泡前6 h內的表面腐蝕形貌均成網狀,結合EDS (圖11h)分析其為Mg(OH)2,浸泡5 d (圖11g)后MZZH試樣的表面出現了片狀的Mg(OH)2。與MZZH相比,MZZMH在SBF中浸泡1 h (圖12a)后基體表面反應形成一層網狀的Mg(OH)2,浸泡1.5 h后,其表面的Mg(OH)2層逐漸被一層顆粒狀物覆蓋(圖12b),隨浸泡時間的延長,顆粒層越來越致密,對圖12d進行EDS分析發現其Ca/P為1.01 (圖12k),推測該物質為CaHPO4。浸泡4 h (圖12e)后MZZMH表面又被Mg(OH)2替代。浸泡6 h后,試樣表面形成尺寸較大的顆粒并逐漸致密(圖12g),結合EDS分析其可能為Ca3(PO4)2 (圖12l),從圖12h看出浸泡1 d后試樣的表面形成了一層細小致密的Ca-P顆粒層,說明試樣在SBF中浸泡1 d左右時,MZZMH的腐蝕層是Mg(OH)2層和Ca-P顆粒層交替生長,而MZZH的腐蝕層是逐漸變厚的Mg(OH)2層,這與圖10c中MZZH試樣腐蝕速率的變化較MZZMH平緩的結果相一致。在SBF中浸泡3和5 d后,MZZMH試樣表面分別被網狀和片狀的Mg(OH)2覆蓋,說明浸泡后期MZZH和MZZMH的表面腐蝕產物主要為Mg(OH)2。
圖11 擠壓態MZZH在SBF中浸泡不同時間的腐蝕形貌及EDS分析
Fig.11 Surface morphologies (a~g) and EDS (h) of MZZH for immersion 1 h (a), 2 h (b), 4 h (c, h), 5 h (d), 6 h (e), 3 d (f) and 5 d (g) in SBF
圖12 擠壓態MZZMH在SBF中浸泡不同時間的腐蝕形貌及EDS分析
Fig.12 Surface morphologies (a~j) and EDS (k, l) of MZZMH for immersion 1 h (a), 1.5 h (b), 2 h (c), 2.5 h (d, k), 4 h (e), 5 h (f), 6 h (g, l), 1 d (h), 3 d (i) and 5 d (j) in the SBF
從實驗結果可知,相比于HA,等量添加的m-HA使復合材料的晶粒細化更加顯著,這可能與m-HA表面MgO的包覆有很大關系。Wang等[28]的研究表明,MgO與α-Mg基體間可以形成半共格界面關系,使得m-HA與α-Mg界面處晶格錯配度減小,二者的浸潤性增加。因此,m-HA在基體中分布更加均勻而且不易團聚,可以作為α-Mg基體異質形核的核心,有效降低非均勻形核功,增大形核率,細化基體晶粒。經過擠壓變形后,MZZMH的晶粒尺寸仍然小于MZZH,這除了鑄態組織遺傳以外,與復合材料MZZMH中m-HA顆粒細小,彌散分布,在熱塑性變形過程中可有效釘扎晶界從而抑制Mg-Zn-Zr合金再結晶晶粒長大也有著必然的聯系。
HA的加入可顯著提高MZZ合金的力學強度,尤其是屈服強度提高了超過30 MPa。其主要原因有2個:首先擠壓態MZZH的平均晶粒尺寸小于擠壓態MZZ,因此根據Hell-Petch公式:
式中,d為材料的晶粒尺寸,σ為材料的屈服強度,σ0反映晶內對變形的阻力,K反映晶界對變形的影響系數,MZZH的屈服強度高于MZZ合金;其次主要是由于在塑性變形過程中,納米HA顆粒可阻礙位錯線和晶界的移動,起到釘扎位錯的作用。對HA納米陶瓷顆粒進行表面改性可顯著提高HA/Mg-Zn-Zr復合材料的硬度和強度。其硬度的提高主要是由于MZZMH中晶粒更細小均勻,晶界密度更高,而晶界處的硬度要大于晶內,所以提高了其硬度。強度的提高主要有以下幾個原因:(1) 由于MZZMH的晶粒尺寸比MZZH小,相應的晶界數量就會增加,高密度的晶界可以更加有效地阻礙位錯滑移系的啟動;(2) 根據Hell-Petch公式,晶粒尺寸更小的MZZMH具有更高的屈服強度;(3) 由于m-HA顆粒在合金基體中的分布均勻彌散,導致MZZMH中增強體粒子間距變小,這對繞過位錯運動的阻力增大,起到明顯的強化作用。此外,由于MgO與α-Mg基體間可以形成半共格界面,因此在塑性變形時由MgO包覆的HA與基體的變形更加協調,所以MZZMH在塑性變形時可承受更大的應力。
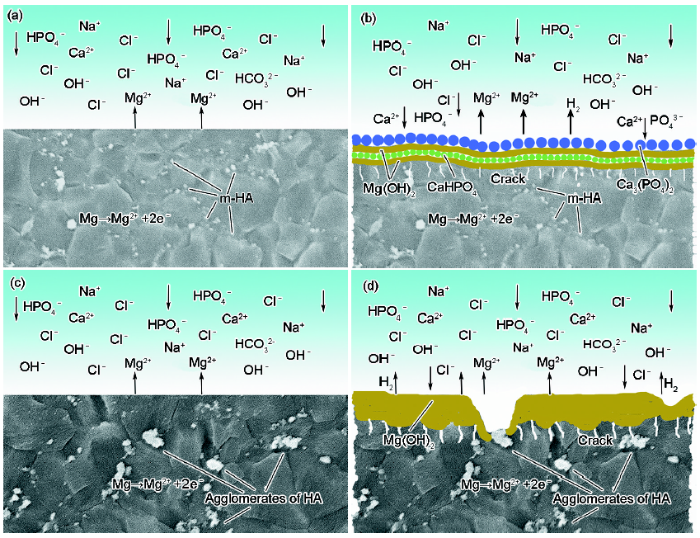
由電化學分析可知,HA的加入可改善MZZ的耐蝕性,這是由于HA的加入可細化晶粒組織,使材料的整體腐蝕更趨近于均勻腐蝕。結合MZZH與MZZMH的電化學測試結果及2種材料浸泡后表面腐蝕產物層的形成過程,不難發現MgO改性HA鎂基復合材料在SBF中初期的腐蝕機理發生了改變,MZZMH和MZZH 2種復合材料在SBF中可能的腐蝕機理如圖13所示。可以看出,在浸泡早期,隨腐蝕時間增加,MZZMH表面Mg(OH)2層和Ca-P顆粒層呈交替生長,相互競爭關系。由于擠壓態MZZMH的晶粒尺寸更加細小均勻,晶界密度高,降低了晶粒與晶界之間的電勢差,使之形成更加密集和微弱的電化學電池區域。同時,高密度的晶界為Mg(OH)2提供了更多的形核位點[29],使表面腐蝕產物Mg(OH)2層相比MZZH更加致密。此外,MZZMH中m-HA顆粒分散均勻、界面結合狀態好,降低了鎂合金基體與增強顆粒之間界面的點陣畸變程度,進而降低界面能,也使MZZMH復合材料的腐蝕敏感性下降。同時,MZZMH復合材料中增強體表面包覆了MgO,當其浸泡于SBF中時,暴露于表面的m-HA會與基體鎂合金一樣與H2O反應生成Mg(OH)2,從而使MZZMH復合材料表面的Mg(OH)2層更加連續、致密,具有較強的保護性,可以有效阻礙SBF對基體的腐蝕。同時,由于Mg(OH)2的生物活性,具有誘導SBF中鈣磷化合物形核生長的作用,而使Ca-P顆粒不斷沉積到Mg(OH)2層的表面,形成圖12中所示的Ca-P顆粒層(圖13b)。顯然,在浸泡初期MZZMH復合材料表面Mg(OH)2層和Ca-P顆粒層相向生長,相互覆蓋,使得腐蝕呈均勻態勢發展,基本抑制了點蝕過程(圖9)。而隨著浸泡時間的不斷增加,在1 d以后,SBF溶液穿透Ca-P顆粒層和Mg(OH)2層腐蝕基體的速率增加,Ca-P顆粒沉積速率相對減緩,已經不足以形成連續的顆粒層,也就失去了保護作用,導致腐蝕層的生長最終以Mg(OH)2層為主。與MZZMH相比,由于MZZH復合材料中HA團聚較為嚴重,其在SBF中浸泡初期的腐蝕產物層Mg(OH)2不僅疏松,而且不連續(在表面HA處斷開),如圖13d所示。MZZH腐蝕層的生長始終以Mg(OH)2為主,MZZH在腐蝕早期的主要形貌為疏松的網狀結構的Mg(OH)2,這是由于MZZH合金內HA的團聚作用導致HA與基體的界面處會出現較為嚴重的腐蝕,且當HA暴露在SBF中時,其與周圍基體形成的高電勢差會形成較嚴重的電偶腐蝕,MZZH與SBF接觸后產生劇烈反應并析出大量H2 (圖10b),H2產生的氣壓會對SBF以及各種離子有一定的排斥作用,Ca-P顆粒無法在疏松且不連續的Mg(OH)2層上沉積成一層保護膜,因此只能生成Mg(OH)2層,這種網狀結構無法有效阻擋SBF,且Mg(OH)2會與Cl-發生反應并被不斷溶解,不利于材料后期的腐蝕。因此,MgO改性HA可顯著改善鎂基復合材料的耐蝕性。
圖13 MZZMH和MZZH在SBF中浸泡前期的腐蝕機理圖
Fig.13 Corrosion mechanism of MZZMH (a, b) and MZZH (c, d) immersed in SBF(a) galvanic corrosion in MZZMH(b) formation of the alternative protective film of Mg(OH)2 and Ca-P particles(c) galvanic corrosion in MZZH(d) formation of Mg(OH)2 layer
(1) HA表面包覆MgO后形成m-HA納米棒,在HA和MgO的界面會有晶格的相互滲透,不是簡單的機械結合,且包覆在HA外面的MgO比較薄。
(2) HA的加入細化了Mg-Zn-Zr合金的晶粒,HA改性后對鑄態及擠壓態HA/Mg-Zn-Zr復合材料的微觀組織具有明顯的細化效果,陶瓷顆粒比較均勻地分布在晶粒和晶界上。
(3) 通過對HA陶瓷納米棒改性,MZZMH復合材料的屈服強度和抗拉強度都得到提高,分別達到291和325 MPa。MZZMH的自腐蝕電位比MZZH高出59 mV。
(4) MZZMH的耐蝕性能優于MZZH。MZZMH的腐蝕速率較MZZH降低,在SBF中浸泡7 d后分別穩定在5和12 mm/a。2種復合材料在SBF中浸泡過程中的腐蝕機理不同。在浸泡前期MZZMH試樣表面腐蝕層以Mg(OH)2層與Ca-P顆粒層交替生長為主,MZZH試樣表面腐蝕層的生長始終以Mg(OH)2為主。
 , 李禎
, 李禎
1 實驗方法
2 實驗結果
2.1 m-HA的表征
2.2 顯微組織


2.3 材料的性能分析
Composite
Ecorr (vs SCE)
icorr
βc (vs SCE)
βa (vs SCE)
Rp
Pi
R1
R2
R3
mV
μAcm-2
mVdecade-1
mVdecade-1
kΩcm2
mma-1
Ωcm2
Ωcm2
Ωcm2
MZZMH
-1403±12
217±14
149±20
126±7
1.37±0.22
4.96±0.32
61.00±2.3
37.18±1.6
7.55±0.9
MZZH
-1462±18
436±17
254±13
176±8
1.04±0.09
9.96±0.39
53.12±1.2
54.14±1.1
6.61±0.3
MZZ
-1542±10
501±13
257±15
219±5
1.03±0.06
11.45±0.29
47.09±3.3
10.05±1.0
-
2.4 MZZH及MZZMH的體外浸泡實驗



3 分析討論
4 結論
來源--金屬學報
 滬公網安備31011202020290號
滬公網安備31011202020290號
