



分享:固溶體中的化學結構單元與合金成分設計

董闖
摘要
工業合金具有特定的牌號成分,理解這些特殊成分背后的結構根源可以從原子結構層面上指導新合金的研發,有效縮短工業合金的制備流程。工業合金多以固溶體結構為基礎,而固溶體以化學近程有序為結構特征,長期以來,人們只能以統計方式獲得溶質元素偏離平均結構的程度,由于缺失描述近程序的精確結構分析方法,導致無法構建能夠指導合金成分設計的有效結構模型。既然優質合金均具有特殊成分,這些成分背后一定對應于類似于分子的特定結構單元。本課題組提出了一種全新的近程有序描述方式——團簇加連接原子。該模型認為,對于固溶體合金,存在理想滿足原子間相互作用的化學結構單元,僅涵蓋第一近鄰團簇以及若干次近鄰的連接原子,可表示為團簇成分式的形式:[團簇](連接原子)。這種團簇式類似于化學物質的分子式,是代表合金平均結構的最小結構單元。通過將Friedel振蕩機制引入到團簇加連接原子模型中,建立了固溶體的團簇共振模型,給出了團簇的球周期近鄰堆垛方式,從而解決了原子密度的關鍵問題。結果表明,團簇成分式中所包含的原子個數正比于體系的平均原子密度和團簇半徑的立方,由此可以定量地計算出理想化學結構單元的具體形式。本文列舉了根據公式計算得到的典型銅基二元合金最佳化學結構單元,計算所得成分與最常用工業合金高度吻合。本工作為成分設計提供了一種新的實用方法。
關鍵詞:
工業合金多以固溶體結構為主,其發展依賴于長時間的經驗探索和冗長的工程實踐,每一種工業合金的研發都需要大量的投入,如何簡化設計流程是人們一直以來關注的問題。成熟的工業合金具有特殊的成分,這表明一定存在某種類似于分子的結構單元,能夠反映工業合金成分的特殊性。長期以來,人們忽視了合金的成分根源這一基礎問題,主要原因是缺少合適的原子結構模型來處理這些固溶體合金。需要說明的是,即便是服役組織復雜的工業合金,其高溫態往往呈單相,例如各種組織形態的鋼均對應于高溫單相奧氏體固態,在這個區間可以實施均勻化處理以彌補鑄態缺陷,高溫母相固溶體的穩定性決定了后續組織演化。因此固溶體的模型化及其穩定性是合金成分設計的普適性基礎。
固溶體合金在長程范圍上保持著溶劑原子的點陣結構,但溶質原子在合金中的化學分布相對復雜,如何理解溶質原子在溶劑原子點陣中的分布模式以及相關的結構穩定性是建立固溶體結構模型的關鍵。對固溶體中原子結構的研究最早可追溯到1919年,Tammann[1]提出經過長期退火的合金其原子傾向于分離并且位于規則的位置。Johannson和Linde[2]通過X射線分析方法證實了在Au-Cu合金中確實存在這樣的分離。Bradly和Jay[3]在Fe-Al體系中也發現了類似現象。以上實驗結果表明合金中的有序程度與溫度密切相關。1934年,Bragg和Williams[4,5]提出了利用長程有序參數S來定量描述固溶體合金中的長程有序結構。隨后Bethe[6]對Bragg-Williams方法進行改進,提出近程有序參數σ (order of neighbors)來描述在一個給定原子附近找到一個不相同原子和找到一個相同原子可能性的差異。直到1950年,Cowley[7,8,9]提出短程序參數(short-range order parameter) α來描述固溶體合金中的短程有序特征。它給出了二元固溶體合金中,以溶質原子為心,每一層的化學成分與平均成分的偏差。然而,α序參數只揭示了圍繞某個溶質的中程范圍內化學元素振蕩分布的圖像,不能給出一個給定固溶體中任何有關化學成分起源的信息,更不能理解具有優異性能的工業合金的成分選取。解決這個基礎而實際的問題需要建立固溶體合金近程序結構模型。
本文首先總結本課題組過去十余年發展的團簇加連接原子模型,該方法適用于描述各種合金中的近程有序結構,包括準晶、非晶和固溶體[10]。該模型認為任何一個合金相的近程序結構都可以簡化成第一近鄰團簇加上若干位于次近鄰的連接原子2部分,團簇相互孤立,其間隙由連接原子填充,表示成統一的團簇式形式為:[團簇](連接原子)x,其中x是連接原子的個數。這種團簇式形式的結構單元被稱做化學結構單元[11]。通過引入Friedel振蕩機制,可解決團簇密堆的中程序堆垛結構以及相應的原子密度計算公式,進而可以定量計算固溶體合金的化學結構單元的成分式。最后,利用該方法計算得到銅基二元體系Cu-(Zn, Ni, Al, Be, Sn)工業合金中的最佳化學結構單元,并且這些成分式與對應體系中最常用的工業合金牌號高度吻合,說明優質合金的特定成分的確源自化學近程序結構單元,從而為理解現有合金成分及進一步合金優化提供了一種全新而有效的方法。
團簇加連接原子模型始于準晶的描述,后來在非晶合金體系得到完善[10],近年來用于合金固溶體[12]。其核心思想為任何結構均可描述為第一近鄰團簇加上若干次近鄰連接原子的單一結構單元,表述為團簇成分式[第一近鄰團簇](連接原子)。以一種常見的非晶合金晶化相Al2NiZr6 (InMg2結構)的結構描述為例,該結構的單胞里含有4種結構占位,以其為心,定義出4種配位多面體團簇,分別為[Ni-Zr9Ni2]、[Al-Zr9Al2]、[Zr1-Ni2Zr8Al2]、[Zr2-Ni1Zr8Al4],這里方括號里面的第一個原子為中心原子,橫線后面為第一近鄰殼層上的原子,上角標表示非等效的原子占位,下角標表示原子的個數。相應的團簇成分式為[Ni-Zr6]Al、[Al-Zr3]Ni1/2、[Zr1-Ni1/3Zr1Al2/3]、[Zr2-Ni1/3Zr1Al2/3]。注意到,第一種團簇式具有最多的原子個數,顯示出該團簇具有最大的孤立度,即團簇特性最為明顯,應該具有最高的結構穩定性,實際上,就是該團簇,搭配以3個連接原子,給出該體系最佳非晶形成能力成分點:[Ni-Zr9Ni2]Al2Ni≈Zr60Ni26.7Al13.3[13],說明了相近結構之間存在著某種特定團簇的結構遺傳,該團簇被稱為該相的主團簇,并認為它構成了對應的非晶態的主體局域團簇[14]。事實上,目前所發現的塊體非晶合金成分均滿足源自對應進行的某種主團簇加上1個或者3個連接原子,且每個團簇式所含價電子個數e/u恒定為24電子[15],例如Cu-Zr體系中具有最高非晶形成能力的成分點之一Cu64Zr36可完美解析為[Cu-Cu7Zr5]Cu≈Cu64.3Zr35.7,e/u≈23.7[16]。需要指出的是,非晶態中的團簇完全孤立,其間由連接原子填充,因此結構和成分式在團簇加連接原子模型下變得極其簡單,正如Macky當年所提出的[17]。
由于上述結構描述方法實際上體現了近程序結構的作用,屬于一般性的結構圖像,類似的團簇式規律應該同樣適用于由化學近程序主導的任何結構。例如Al6Fe準晶的團簇式為[Fe-Fe1Al11]Al≈Al85.7Fe14.3≈Al6Fe,這里的二十面體團簇源自晶化相Al3Fe[18]。下面介紹工業合金的基礎結構——固溶體的結構特征以及團簇模型。
合金固溶體以化學近程序為特征,因此早期的固溶體結構模型[19,20]一般基于短程有序或者電子結構。對于前一種情況,溶質原子和溶劑原子根據它們之間的相互作用模式表現出近鄰構型,溶質原子在溶劑原子基體點陣中的徑向分布可以用序參數來描述。對于后一種情況,忽略單個電子的行為,把合金看成原子和價電子的混合,其穩定機制由Fermi面和Brillouin區相互作用決定[21],認為當Fermi面和Brillouin區相切的時候系統最穩定,即2kF≈Kp (kF為Fermi波矢,Kp為Brillouin區的寬度)。Friedel給出了這種情況下的嚴格處理方法,提出Friedel振蕩描述固溶體結構[22],指出雜質電荷(這里為溶質原子)周圍的屏蔽電子會產生極化,從而導致電子密度的振蕩分布。這種強烈的電荷屏蔽作用發生在溶質原子的周圍,是短程有序形成的根本原因。
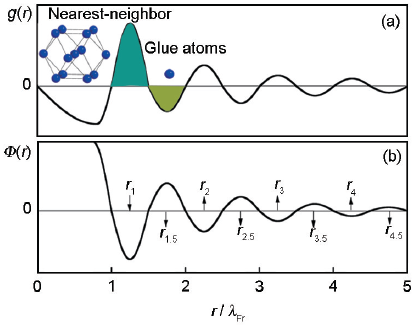
金屬中含有大量的傳導電子,當一個雜質電荷被引入到電子體系時,雜質電荷周圍的電子云會產生極化來屏蔽雜質電荷對體系產生的擾動,導致雜質電荷周圍電子云密度的重新分布[23]。雜質周圍屏蔽電荷表現出的這種長程振蕩就是所謂的Friedel振蕩[24]。如圖1所示,在液相和金屬玻璃中,一個位于r處的電子能夠感受到的總勢能函數Φ (r)正比于一個正弦函數,Φ (r)∝-sin(2kFr)/r3,勢函數形式如圖1b所示。電子的這種振蕩行為會反過來影響原子密度的分布,引起正空間原子密度的振蕩。圖1a所示為對分布函數g(r),可以看出原子密度在勢函數波谷處取得極大值,即原子傾向于占據勢函數波谷的位置。勢函數波谷的中點位于rn=(1/4+n)λFr,n=1、2、3、…,其中λFr=π/kF,為Friedel波長。液相和金屬玻璃中原子的這種層狀分布稱為球周期序。這些勢函數的波谷區域用rn表示,波峰區域用rn+0.5表示,如圖1b所示。勢函數第一個波谷的中心位置r1對應于團簇半徑r1=(5/4)λFr=1.25λFr。
圖1 對分布函數g(r)及Friedel振蕩的有效對勢函數Φ(r)∝-sin(2kFr)/r3
Fig.1 Pair distribution function g(r), with the location zones of the nearest-neighbor and glue atoms being labeled (a), and the corresponding effective electronic potential Φ(r)∝-sin(2kFr)/r3 arising from Friedel oscillation (b) (rn and rn+0.5 mark respectively the central positions of the negative and positive potential zones; the radial distance r is scaled with Friedel wavelength λFr)
依據團簇加連接原子模型,對上述結構圖像做如下簡化:
(1) 中程范圍上的振蕩可以被簡化為一個最小的區域,在這個區域內,電荷的擾動接近中性,對應于最近鄰的團簇部分加上位于團簇外層的連接原子部分,從而定義出固溶體中的類似于分子的化學結構單元。
金屬中電子的這種不完全屏蔽行為表明由中心雜質(溶質原子)產生的電荷振蕩不能被最近鄰原子完全屏蔽(與分子中第一近鄰形成完全電中性對比)。因此,在勢函數中一個深的波谷區域r1外,總會伴隨著一個波峰區域r1.5,隨后又會有一個波谷區域r2。只有到了一定的中程距離之后,振蕩完全衰減,體系呈現電中性。由于勢函數以距離的三次方形式迅速衰減,其中第一個波谷和第一個波峰的振蕩相對最強烈。由此,依據本課題組前期發展起來的團簇加連接原子模型[10],定義一個類似于分子的化學結構單元(chemical unit),涵蓋位于r1的最近鄰團簇部分加上位于r1.5的連接原子部分,這是近程序結構的最極致的簡化,如圖1a中陰影部分所示。在理想滿足原子間相互作用情形下,這種簡化在r1和 r1.5區內接近于電中性,雜質電荷的影響在第一個振蕩周期內基本得到完全屏蔽。這就是高穩定性合金必然具有簡單團簇成分式的根本原因,即化學結構單元就是描述理想近程序結構的團簇式。化學結構單元就是合金中的“分子”,兼具結構、成分和電子結構信息,其成分由類似于分子式的團簇式描述。
對于稀固溶體,由于溶質的含量非常少,2個溶質所在的團簇式相距非常地遠,溶質間的相互作用幾乎可以忽略。對于濃固溶體,也就是本工作所討論的系統,溶質含量較高,溶質所在的團簇式之間存在關聯,需要明確團簇式之間的堆垛模式,建立完整的結構模型。
(2) 結合中程范圍的球周期性,建立團簇共振模型,給出化學結構單元之間的堆垛方式,最近鄰團簇位于勢函數第三個波谷的位置r3=(13/4)λFr=2.6r1。
近鄰原子之間形成團簇是原子層面上的近程有序,而在中程序上團簇和團簇之間還存在著一定的有序性,可以視為球周期性的延伸,表現為相鄰的團簇傾向位于勢函數第三個波谷處r3的位置[15]。團簇的半徑為r1=1.25λFr,在沒有團簇搭接的情況下(即團簇保持完全的孤立性,類似于非晶態中主團簇的分布行為),2個相鄰團簇之間的距離為2.5λFr。如果相鄰團簇位于勢函數第二個波谷r2區域(2~2.5λFr),近鄰團簇與中心團簇之間將會產生搭接,從而破壞化學結構單元的完整性。因此,近鄰團簇傾向位于勢函數的第三個波谷r3區域(3~3.5λFr)。以fcc純金屬結構為例說明化學結構單元的堆垛方式。fcc結構中的團簇為CN12 (coordination number)的立方八面體團簇[25],中心原子到最鄰原子的距離,也就是團簇半徑為r1=<1/2, 1/2, 0>a=
由于固溶體中團簇式的定義以及分布與非晶態中主團簇的完全一致,因此,固溶體可以視為化學非晶態(chemical amorphous state),即在給定的基礎點陣架構上,溶質元素以類似于非晶態的團簇形式散布于溶劑的基體上。
圖2 fcc結構中最理想的球周期堆垛方式
Fig.2 Ideal spherical-periodicity packing of clusters in fcc structure, where neighboring cuboctahedral clusters are located at <3/2, 1, 1/2>a position with the distance of r3=2.6r1, a being the lattice constant of the base fcc lattice
在上節建立了一個完整的結構模型,包括描述近程序的化學結構單元以及結構單元在中程序上的堆垛。明確結構單元在中程上的堆垛方式可以計算出濃固溶體中單位化學結構單元中包含的總原子個數,實現連接原子個數的準確定義。
建立了固溶體結構的結構模型之后,利用該模型可計算高溶質含量的置換型固溶體中的化學結構單元,這些固溶體滿足單一化學結構單元,并且團簇之間滿足球周期排列,相鄰團簇位于r3區域。
首先計算固溶體的平均原子密度。假設團簇加連接原子模型的局域有序和團簇堆垛的中程球周期序在近熔點狀態的液體和其對應的初級固溶體中保持不變。這個短程序結構遺傳性是一個合理的假設,例如,中子衍射實驗[26]表明,液態Fe接近熔點時的近程序與高溫結構δ-Fe中的近程序非常相似;液態Al在熔點附近的X射線衍射實驗[27]表明,其結構因子與對應晶態中的接近。
非晶合金的團簇共振模型[15]指出,具有高形成能力的理想金屬玻璃(這里稱為高液態穩定性),其團簇之間的堆垛滿足球周期序,相鄰團簇位于Friedel振蕩勢函數的第三個最小值處,r3=2.6r1。假設團簇是由硬球堆垛而成,連接原子位于團簇的間隙,平均原子密度ρa可以通過計算r3為半徑的體積內所包含原子的個數來確定,并且ρa與團簇式內含有的原子個數Z有關:ρa=[13.33×(13/32)+1]×Z/(r33×4π/3)。因此一個化學結構單元中含有的總原子個數Z可以表示為:
式中,c為常數。當最近鄰團簇恰好位于勢函數第三個波谷的中點r3=2.6r1時,此時團簇排布滿足理想共振態,c=(4π/3)×2.63/(13.33×13/32+1)≈11.476。對于非理想排布的團簇,近鄰團簇位于r3的區域內,這個區域的邊界為2.4r1和2.8r1,此時常數c的下限和上限分別為9.026和14.333。
對于純金屬可以直接算出Z值,因為ρar13為常數。例如,對于fcc結構金屬,ρa=4/a3 (每個單胞中有4個原子,a為晶格常數),團簇半徑為r1=
對于固溶體合金來講,平均原子密度ρa和團簇半徑r1都取決于溶質原子在團簇式中的位置,而這個位置取決于溶質原子與溶劑原子相互作用的模式:
(1) 相互吸引。這種情況通常發生在2種原子混合焓ΔH為負值的時候,即ΔH<0 (ΔH表示合金體系中異類原子成鍵與同類原子成鍵時系統勢能的差異。ΔH<0,表示異類原子成鍵時勢能更低,系統更穩定;ΔH>0,表示系統傾向于同類原子成鍵)。溶質原子傾向于位于團簇的中心,這樣才能與周圍的溶劑原子產生最大的相互作用,形成溶質原子為中心、溶劑原子為殼層的團簇結構。當溶質原子的含量過多時,多余的溶質將占據連接原子的位置。例如,fcc結構的Cu-Zn固溶體,2種原子的ΔH<0[28],且溶質Zn原子的含量過多,理想的團簇部分為[Zn-Cu12],連接原子部分為Cu和Zn的混合。
(2) 相互排斥。這種情況通常發生在ΔH>0的時候。溶劑原子傾向于與溶質原子分離,團簇部分由溶劑原子構成,溶質原子則更傾向于占據連接原子的位置。例如,對于fcc結構Cu-Ni固溶體,混合焓為正值ΔH=+2 kJ/mol[29],理想的團簇部分為[Cu-Cu12],連接原子為Cu和Ni的混合。
(3) 中性作用。這種合金系統中的溶質原子與溶劑原子的性質非常接近,因此溶質原子可以形成近乎隨機分布的模式。盡管在真實的合金中很少有這樣的例子,這種類型的固溶體可以被看作由平均原子構成,因此化學結構單元與純金屬中的情況一致,例如對于fcc結構,其形式為[M-M12]M3。
對于fcc結構固溶體,溶劑原子B構成單層的立方八面體團簇,團簇半徑r1=2RB,RB為溶劑原子B的半徑;當溶質原子A占據團簇中心時,團簇半徑變為r1=RA+RB,RA為溶質原子A的半徑。
平均原子密度ρa定義為單位體積內包含的原子的個數,是平均原子體積Va的倒數。Va根據所有溶質原子和溶劑原子的原子體積分數之和計算獲得:1/ρa=Va=ΣfiVi,fi和Vi分別為元素i的原子分數和原子體積。這個計算是建立在2種元素混合時原子體積保持不變的基礎上。對于fcc結構固溶體,Vi=(4π/3)Ri3/0.74,0.74為fcc結構的密堆率,Ri為元素i的原子半徑。
因此對于任意fcc結構的濃固溶體系統A-B,A為溶質原子,B為溶劑原子,理想的化學結構單元可以表示為[A-B12]AxBy或[B-B12]AxBy。對于第一種情況,式(1)變為:
由Z=13+x+y,(x, y)的關系可以表示為:
式中,RA/B為溶質原子和溶劑原子的半徑比。
對于第二種情況,溶質原子只占據連接原子位置,公式(1)中的團簇半徑r1等于2RB,[B-B12]AxBy中的(x, y)表達式為:
對于式(3)和(4),最接近整數解的(x, y),只與溶質原子相對溶劑原子的半徑比RA/B有關。
本節利用固溶體結構模型分析fcc結構典型銅基二元工業合金中的化學結構單元,包括Cu-Zn、Cu-Ni、Cu-Al、Cu-Be和Cu-Sn,這些體系的相圖中有相對較寬的固溶區間,并且溶質的濃度較高,能夠用單一的化學結構單元來描述。解析后的化學結構單元列于表1。
表1 Cu基工業合金的最佳化學結構單元
Table 1 Optimum chemical units of Cu-based commercial alloys
在高溫下,fcc結構的銅基體中可以固溶39% (質量分數,下同)的Zn[30]。根據ASM手冊[31],α黃銅具有較寬的的成分區間,從Zn含量最低的合金牌號C21000 (95Cu-5Zn)一直到Zn含量最高的Muntz合金C28000 (60Cu-40Zn)。在這些牌號之中,彈殼合金C26000 (70Cu-30Zn)的應用最為廣泛。
很早以前就有人仔細測量過α黃銅單晶樣品中的Cowley短程序參數α[32]。以溶質原子Zn為中心,最近鄰殼層的α值為α110=-0.1373,負值表明第一近鄰傾向于異類原子Zn-Cu近鄰,下一殼層的α值為α200=0.1490,表示該殼層傾向于Zn-Zn近鄰,這種分布方式滿足Zn-Cu之間負混合焓所預期的情況。根據團簇加連接原子模型,當合金中含有足夠的Zn時,化學結構單元可以表示為[Zn-Cu12]ZnxCuy,連接原子是Zn和Cu的混合。這種表達形式與實驗結果一致,Zn原子為心,第一近鄰為12個Cu原子,表現為Zn-Cu近鄰,與第一殼層為負的序參數α110吻合;連接原子中為Zn和Cu的混合并且傾向于Zn原子富集,這樣才能表現出Zn-Zn近鄰的趨勢,與第二殼層為正的序參數α200吻合。根據式(2),化學結構單元中包含的總原子個數Z與Zn和Cu之間的原子半徑差異有關。對于金屬原子采用十二配位多面體情況下的Goldschmidt半徑,RA=RZn=0.139 nm和RB=RCu=0.128 nm[33]。通過計算獲得x和y之間的關系式滿足1.28x+y=5.12。這個關系式所給出的連接原子中Zn、Cu原子的個數具有多種組合情況,每一種情況對應的化學結構單元都同時滿足局域近程序和中程球周期序。
1.28x+y=5.12最接近整數的(x, y)解為(4, 0),即連接原子位置完全被4個Zn原子占據,形成了[Zn-Cu12]Zn4形式的化學結構單元,在這種情況下,最近鄰的團簇最接近于中r3的位置,滿足理想的球周期分布。[Zn-Cu12]Zn4=70.0Cu-30.0Zn (%),恰好為使用最廣泛的彈殼黃銅合金C26000 (70Cu-30Zn)。
Cu-Ni合金由于其優異的抗海水腐蝕能力被廣泛應用在管道和船舶材料中。Cu-Ni合金的工業牌號也有著較寬的成分范圍,從Ni含量最低的C70400 (95Cu-5Ni)到Ni含量最高的C71500 (70Cu-30Ni)。最常用的牌號為C70600 (90Cu-10Ni)和C71500 (70Cu-30Ni)[31]。
由于Cu-Ni之間的正混合焓ΔHCu-Ni=+2 kJ/mol[29],Ni原子傾向于占據團簇式中連接原子的位置,團簇式形式為[Cu-Cu12]NixCuy。Ni原子的Goldschmidt半徑為RA=0.125 nm[33]。根據式(3),得到(x, y)的關系式0.93x+y=3.22。這個關系式沒有非常接近整數值的(x, y)解,因此需要考慮所有x+y=3的組合。團簇式形式為[Cu-Cu12](Ni, Cu)3,意味著連接原子占位上的Ni原子和Cu原子傾向于隨機分布。這一點可以通過實驗結果證實:在Cu80Ni20合金中,第二殼層的短程序參數值非常小,α2=α200=-0.058[34],這個值大約只有Cu-Zn合金的1/3,表示該位置只有輕微富Ni的傾向。考慮到這一點以及x+y=3,最可能的團簇式形式為[Cu-Cu12]Ni2Cu1=88.3Cu-11.7Ni (%),該團簇式解釋了最常用的合金牌號C70600 (90Cu-10Ni)。另外2個整數解對應的團簇式形式[Cu-Cu12]Ni1Cu2和[Cu-Cu12]Ni3沒有對應的合金牌號,因為不滿足連接原子位置上Ni原子只是輕微富集的事實。
Cu-Al合金有高強度、高的抗氧化性和耐機械磨損性。典型的Cu-Al二元工業合金只有2個:C60600 (95Cu-5Al)和C60800 (92Cu-8Al)[31]。Al在Cu中的最大固溶度為9.3%[35],因此Cu-Al合金的牌號相對較少。
Cu-Al的混合焓為ΔHCu-Al=-1 kJ/mol[36],Al原子傾向于占據團簇中心的位置,對應的團簇式形式為[Al-Cu12]AlxCuy。Al原子的Goldschmidt半徑為RA=0.143 nm[33]。根據式(2),得到(x, y)的關系式1.39x+y=5.85。最接近整數的(x, y)解為(2, 3),對應的團簇式形式為[Al-Cu12]Al2Cu3=92.2Cu-7.8Al (%),很好地解釋了合金牌號C60800 (92Cu-8Al)。另一個合金牌號C60600 (95Cu-5Al)可以暫時利用團簇式[Al-Cu12]Al1Cu4=95.0Cu-5.0Al (%)來解析,這時對應的(x, y)解為(1.3, 4),偏離整數解1。這2個合金牌號中,92Cu-8Al的原子在近程上和團簇在中程上的堆垛更接近于理想球周期序。
Cu-Be合金具有高強度和高彈性,可以作為好的熱電導體,最常用的合金牌號為C17200 (98Cu-2Be)[31]。
Cu-Be之間的混合焓接近于0[36],表明Cu和Be之間的相互作用較弱,相對于之前分析的合金而言,該體系中的溶質原子傾向于隨機分布。實驗測得的短程序參數為正[37],這表明Cu-Be之間傾向于彼此分離,一般的化學結構單元形式為[Cu-Cu12]BexCuy。Be原子的Goldschmidt半徑為RA=0.113 nm[33]。根據式(3),得到(x, y)的關系式為0.69x+y=3.22。最接近整數的(x, y)解為(2, 2),對應的團簇式為[Cu-Cu12]Be2Cu2=98.1Cu-1.9Be (%),恰好對應于最常用的合金牌號C17200 (98Cu-2Be)。Be在Cu中的最大固溶度為2.7%[38],這個成分位于固溶極限內。
錫青銅是一種非常古老的合金,有很多合金牌號,但是最常用的合金牌號為C90800 (88Cu-12Sn)[31]。
Cu-Sn的混合焓ΔHCu-Sn=+7 kJ/mol[36],Sn原子傾向于占據連接原子的位置,一般的團簇式形式可以表示為[Cu-Cu12]SnxCuy。Sn原子的Goldschmidt半徑為RA=0.155 nm[33]。根據式(3),得到(x, y)的關系式1.78x+y=3.22。唯一最接近于整數的(x, y)解為(1, 1),對應的團簇式形式為[Cu-Cu12]Sn1Cu1=88.2Cu-11.8Sn (%),很好地解釋了最常用的合金牌號C90800 (88Cu-12Sn)。
本文總結了基于團簇加連接原子模型的固溶體近程有序結構描述以及相關的類分子團簇成分式。將Friedel振蕩機制引入到團簇加連接原子模型中,建立了固溶體的電子結構模型,提出化學結構單元的概念,可以看作是合金中的分子式,是最小的電中性結構單元,用團簇加連接原子模型表示為團簇部分加上位于團簇間隙的連接原子部分,分別對應Friedel振蕩勢函數中的第一個波谷r1和第一個波峰r1.5位置。團簇之間沒有搭接并且滿足球周期序堆垛,相鄰團簇位于Friedel振蕩勢函數的第三個波谷r3處。通過建立團簇在中程上的堆垛方式,給出了每個結構單元所包含原子個數Z的計算公式,Z與體系的平均原子密度ρa和團簇半徑r1呈簡單的正比例關系,Z=cρar13。通過計算,獲得了最常用的二元銅基工業合金的化學結構單元,包括[Zn-Cu12]Zn4、[Cu-Cu12]Ni2Cu1、[Al-Cu12]Al2Cu3、[Cu-Cu12]Be2Cu2和[Cu-Cu12]Sn1Cu1,對應合金牌號分別為C26000 (70Cu-30Zn)、C70600 (90Cu-10Ni)、C60800 (92Cu-8Al)、C17200 (98Cu-2Be)和C90800 (88Cu-12Sn),是各體系中公認的性能最優的合金牌號。
 , 董丹丹
, 董丹丹
1 固溶體合金的特征化學近程序結構描述:團簇加連接原子模型及化學結構單元
2 化學結構單元的計算
3 典型銅基二元工業合金的化學結構單元
System
ΔH
Cluster
Glue atoms AxBy
Composition
Composition
Alloy specification
kJmol-1
formula
%
Cu-Zn
-6
[Zn-Cu12]
1.28x+y=5.12
[Zn-Cu12]Zn4
70.0Cu-30.0Zn
C26000 70Cu-30Zn
Cu-Ni
+2
[Cu-Cu12]
0.93x+y=3.22
[Cu-Cu12]Ni2Cu1
88.3Cu-11.7Ni
C70600 90Cu-10Ni
Cu-Al
-1
[Al-Cu12]
1.39x+y=5.85
[Al-Cu12]Al2Cu3
92.2Cu-7.8Al
C60800 92Cu-8Al
Cu-Be
0
[Cu-Cu12]
0.69x+y=3.22
[Cu-Cu12]Be2Cu2
98.1Cu-1.9Be
C17200 98Cu-2Be
Cu-Sn
+7
[Cu-Cu12]
1.78x+y=3.22
[Cu-Cu12]Sn1Cu1
88.2Cu-11.8Sn
C90800 88Cu-12Sn
3.1 Cu-Zn
3.2 Cu-Ni
3.3 Cu-Al
3.4 Cu-Be
3.5 Cu-Sn
4 結論
來源--金屬學報
 滬公網安備31011202020290號
滬公網安備31011202020290號
