
檢檢測(cè)")

關(guān)注")
分享:γ-APS接枝環(huán)氧樹脂分子對(duì)環(huán)氧涂層/金屬界面化學(xué)鍵合的研究

操發(fā)春
摘要
利用γ-氨基丙基三甲氧基硅烷(γ-APS)對(duì)環(huán)氧樹脂分子進(jìn)行改性合成了具有鍵合特性的活性樹脂(γ-APS/EP),將活性樹脂作為液態(tài)添加劑加入到環(huán)氧涂層中,利用活性樹脂與金屬表面羥基反應(yīng)生成耐水解的噁烷鍵的特點(diǎn),實(shí)現(xiàn)了環(huán)氧涂層/金屬界面的化學(xué)鍵合。研究表明,活性樹脂添加量為3% (質(zhì)量分?jǐn)?shù))時(shí),環(huán)氧涂層與金屬基體的界面結(jié)合力和對(duì)腐蝕介質(zhì)的抗?jié)B透性能達(dá)到最佳。根據(jù)活性樹脂含量的影響,提出了活性樹脂在環(huán)氧涂層/金屬界面的鍵合機(jī)理。
關(guān)鍵詞:
環(huán)氧涂層廣泛應(yīng)用于金屬的海洋腐蝕防護(hù)領(lǐng)域,但在服役過程中,由于海水中的O2分子、Cl-會(huì)伴隨著海水通過涂層中的微觀通道(缺陷、自由體積)逐漸向金屬基體傳輸,引發(fā)溶脹、鼓泡、陰極剝離等問題,最終導(dǎo)致涂層失效[1,2,3,4]。研究[4]發(fā)現(xiàn),涂層與金屬界面的濕態(tài)附著力是決定有機(jī)涂層防腐蝕性能的決定性因素之一。然而,當(dāng)前環(huán)氧涂層與金屬界面的結(jié)合多為物理吸附,界面結(jié)合力弱直接限制了涂層防護(hù)性能的發(fā)揮。
有機(jī)硅烷(簡(jiǎn)稱硅烷),通式為R'(CH2)nSi(OR)3 (R'為有機(jī)官能團(tuán),OR為可水解的烷氧基團(tuán),如—OCH3、—OC2H5等),是一種常用的偶聯(lián)劑,被大量用于復(fù)合材料中以提高強(qiáng)化相(如玻璃鱗片、黏土)與樹脂基體的黏結(jié)力[5]。一般認(rèn)為,硅烷的有機(jī)端與聚合物相容性好,可與聚合物中某些特定基團(tuán)反應(yīng)生成共價(jià)鍵,同時(shí),硅烷無機(jī)端OR易水解生成硅羥基(Si—OH),它既可以縮聚生成硅氧烷,又可以與無機(jī)物表面的羥基進(jìn)行縮合反應(yīng),在界面處生成耐水解的噁烷鍵(Si—O—M,M為無機(jī)原子)。硅烷在界面處如同一個(gè)橋梁,通過化學(xué)鍵合的方式將無機(jī)材料與有機(jī)物連接起來[5,6]。因此,眾多研究人員將硅烷應(yīng)用于有機(jī)防護(hù)涂層領(lǐng)域中(前處理劑、添加劑等),證實(shí)了硅烷能夠提高涂層的濕態(tài)附著力和防腐蝕性能[7,8,9,10,11]。
有機(jī)端為氨基的硅烷是環(huán)氧樹脂防腐涂層體系中最常用的硅烷之一,這是由于氨基能夠與環(huán)氧樹脂中環(huán)氧基團(tuán)反應(yīng)使兩者保持良好的相容性,但是氨基的存在也帶來了一些問題:(1) 氨基的質(zhì)子化。Pefrunin等[12]發(fā)現(xiàn)γ-氨基丙基三甲氧基硅烷(γ-APS)中的氨基會(huì)質(zhì)子化生成NH3+,使膜層呈現(xiàn)電正性,吸引Cl-向金屬界面遷移引發(fā)金屬基體局部腐蝕;(2) 氨基的競(jìng)爭(zhēng)吸附。研究[13,14]發(fā)現(xiàn),當(dāng)氨基硅烷應(yīng)用在鋼表面時(shí),由于氨基的存在,這類有機(jī)硅烷在鋼表面存在2種競(jìng)爭(zhēng)吸附形式:一種是氨基與金屬表面的羥基通過不穩(wěn)定的氫鍵吸附,水存在下易解吸,引發(fā)服役過程中膜層的剝離和溶解;另一種是有機(jī)硅烷中的硅羥基與金屬表面的羥基通過氫鍵吸附,高溫烘干脫水轉(zhuǎn)變成噁烷鍵。耐水解的噁烷鍵是提高涂層附著力的關(guān)鍵,而氨基與硅羥基的競(jìng)爭(zhēng)吸附將導(dǎo)致噁烷鍵的數(shù)目減少。Harun等[6]使用X射線光電子能譜(XPS)檢測(cè)了由γ-氨基丙基三乙氧基硅烷處理的低碳鋼表面,發(fā)現(xiàn)有大量硅烷分子的氨基端朝向金屬基底而無機(jī)端朝向表面,這加速了環(huán)氧樹脂涂層濕態(tài)附著力的喪失。因此,金屬腐蝕與防護(hù)領(lǐng)域在應(yīng)用有機(jī)端為氨基基團(tuán)的硅烷時(shí),必須設(shè)法消除氨基的負(fù)面作用。
本工作提出了一種實(shí)現(xiàn)環(huán)氧涂層/金屬界面化學(xué)鍵合的方法,即利用γ-APS中的氨基和環(huán)氧樹脂中的環(huán)氧基進(jìn)行開環(huán)反應(yīng)合成活性樹脂(γ-APS/EP),將活性樹脂作為添加劑加入環(huán)氧樹脂中參與成膜。該方法一方面利用開環(huán)反應(yīng)避開了有機(jī)硅烷中氨基質(zhì)子化和競(jìng)爭(zhēng)吸附的問題,另一方面充分發(fā)揮有機(jī)硅烷的橋梁作用,利用有機(jī)硅烷與金屬界面羥基反應(yīng)生成耐水解的噁烷鍵,實(shí)現(xiàn)了環(huán)氧涂層/金屬界面的化學(xué)鍵合,并提出了活性樹脂實(shí)現(xiàn)環(huán)氧涂層/金屬界面的化學(xué)鍵合機(jī)理模型。
環(huán)氧涂層試樣以Q235鋼板(50 mm×50 mm×2 mm)作為基底,鋼板在涂刷成膜前需用砂紙逐級(jí)打磨至400號(hào),再依次使用沾有乙醇和丙酮的脫脂棉進(jìn)行擦拭除油,最后用吹風(fēng)機(jī)吹干,置于干燥箱中待用。吸水率測(cè)試以蓋玻片為基底。
以γ-APS和環(huán)氧樹脂E51為本次實(shí)驗(yàn)使用的合成原料。由于γ-APS易水解,所以在合成反應(yīng)前,需要對(duì)環(huán)氧樹脂進(jìn)行脫水處理。基于共沸原理,首先將6 g環(huán)氧樹脂與65 g二甲苯混勻加入錐形瓶中,連接上冷凝管和分水器后,通Ar氣排出空氣。隨后在140 ℃油浴中加熱,在磁力攪拌作用下脫水。
將脫水處理后的環(huán)氧樹脂與γ-APS按摩爾比2∶1加入到錐形瓶中,先在室溫下劇烈攪拌1 h,隨后在60 ℃油浴中攪拌3 h。然后,減壓蒸餾除去二甲苯,得到合成產(chǎn)物,即活性樹脂(γ-APS/EP)。為了防止活性樹脂受潮變質(zhì),將其放在真空干燥器中保存。
試樣制備分為2個(gè)環(huán)節(jié),制備條件:濕度(RH) 60%±5%,室溫。(1) 涂料的制備,使用設(shè)備為SFJ-400多功能攪拌機(jī)。將γ-APS/EP與環(huán)氧樹脂E44按一定比例混合,1500 r/min下攪拌30 min后,將固化劑LITE558按環(huán)氧樹脂E44質(zhì)量的40%加入,攪拌5 min (1200 r/min)后靜置待用。(2) 涂層的制備,分別使用刷涂法和滴涂法在Q235鋼板和蓋玻片上涂膜,膜層在室溫下放置2 h,在60 ℃烘箱中恒溫12 h。Q235試樣涂層厚度控制在(40±10) μm,蓋玻片試樣涂層約為(130±10) μm。所選用的活性樹脂占環(huán)氧樹脂的質(zhì)量分?jǐn)?shù)分別為0%、0.5%、1%、3%、5%和10%,對(duì)應(yīng)的涂層分別稱為清漆(pure epoxy)、0.5%γ-APS/EP、1%γ-APS/EP、3%γ-APS/EP、5%γ-APS/EP和10%γ-APS/EP。
使用重量法測(cè)試計(jì)算涂層體系在3.5%NaCl溶液中的吸水率。吸水率(Qt)計(jì)算公式如式(1)所示,取3個(gè)試樣測(cè)試結(jié)果的平均值為某一時(shí)刻的吸水率:
式中,m0為試樣初始重量,m1為蓋玻片重量,mt為t時(shí)刻試樣的重量。
利用PosiTest AT附著力試驗(yàn)儀根據(jù)ISO4624標(biāo)準(zhǔn)進(jìn)行附著力測(cè)試。Al錠直徑為20 mm,黏合劑為SGA-39快固膠黏劑,加載速率0.70 MPa/s。對(duì)于濕態(tài)附著力,涂層浸泡后先用酒精擦拭干凈,吹干后進(jìn)行附著力測(cè)試。取3~5個(gè)點(diǎn)的附著力測(cè)試平均值作圖。
采用Spectrum Two紅外光譜儀(FTIR)測(cè)試樣品FTIR譜,掃描累計(jì)次數(shù)10次,分辨率為4 cm-1。采用JSM-6301F掃描電子顯微鏡(SEM)和Escalab 250Xi XPS對(duì)樣品進(jìn)行組織觀察和成分分析。其中,SEM工作電壓為10 kV;XPS以AlKα射線為光源,全譜測(cè)試通能為100 eV,步長(zhǎng)1 eV;精細(xì)譜測(cè)試通能為50 eV,步長(zhǎng)0.05 eV,并采用C—C單鍵的C1s (284.6 eV)對(duì)譜圖進(jìn)行電荷校正。采用TA-Q20差示掃描量熱儀(DSC)對(duì)樣品進(jìn)行熱分析,測(cè)試采用ISO 11357-2標(biāo)準(zhǔn),N2氣氛,升溫范圍為20~200 ℃,升溫速率為10 K/min。以Ti,g (即DSC曲線一階微分最大值)為玻璃化轉(zhuǎn)變溫度(Tg)。
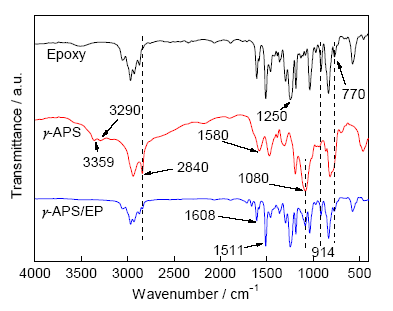
圖1為環(huán)氧樹脂E51、硅烷偶聯(lián)劑γ-APS和活性樹脂γ-APS/EP的FTIR譜。在γ-APS的FTIR曲線中,3359、3290和1580 cm-1 3處是伯氨基(—NH2)的特征振動(dòng)峰[15,16],1192、1082和2840 cm-1是代表甲氧基硅(Si—O—CH3)基團(tuán)的振動(dòng)峰[17]。在環(huán)氧樹脂E51的FTIR譜中,由環(huán)氧基團(tuán)引起的特征振動(dòng)峰有1250、914和770 cm-1,由環(huán)氧樹脂分子中的苯環(huán)引起的振動(dòng)峰有1608、1511和828 cm-1 [18]。與硅烷偶聯(lián)劑和環(huán)氧樹脂對(duì)比,γ-APS/EP的紅外光譜圖主要表現(xiàn)出以下特點(diǎn):一方面,代表環(huán)氧基團(tuán)振動(dòng)的特征峰(如914 and 770 cm-1)的強(qiáng)度在接枝KH540后明顯減弱,說明反應(yīng)消耗了部分環(huán)氧基團(tuán);另一方面,活性樹脂分子FTIR曲線中氨基振動(dòng)峰消失,意味著氨基在反應(yīng)過程中消耗殆盡。這說明接枝反應(yīng)主要是通過環(huán)氧樹脂的環(huán)氧基團(tuán)與γ-APS的氨基的親核加成反應(yīng),合成反應(yīng)的機(jī)理如圖2所示。圖1中1080 cm-1處甲氧基硅基團(tuán)的特征振動(dòng)峰依舊很明顯,說明由于反應(yīng)過程中的良好惰性氛圍保護(hù),有效防止了有機(jī)硅烷的水解。這保證了在后期涂層制備中,活性樹脂能夠水解生成硅羥基繼而與金屬表面羥基進(jìn)行縮合反應(yīng),以達(dá)到涂層與金屬化學(xué)鍵合的目的。
圖1 環(huán)氧樹脂E51、硅烷KH540和活性樹脂γ-APS/EP的FTIR譜
Fig.1 FTIR spectra of epoxy resin, γ-APS and γ-APS/EP
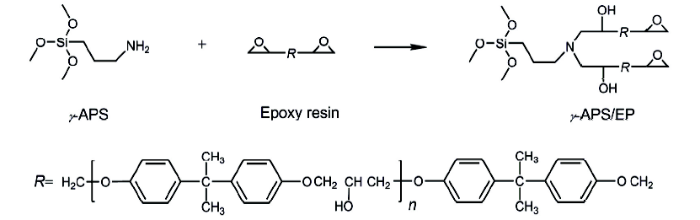
圖2 活性樹脂的合成反應(yīng)機(jī)理示意圖
Fig.2 Schematic of γ-APS/EP preparation
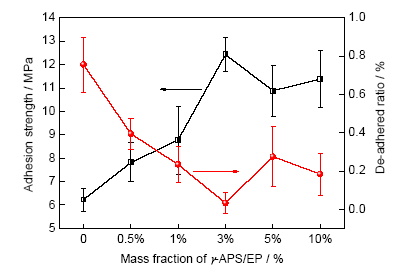
環(huán)氧涂層干態(tài)附著力和剝離率隨活性樹脂含量的變化曲線如圖3所示。可以看出,隨著γ-APS/EP的含量增加,涂層附著力從清漆的6.2 MPa逐漸增加到含量為3%時(shí)的12.4 MPa,而當(dāng)含量進(jìn)一步升高,附著力反而略有下降。涂層剝離率是附著力拉拔測(cè)試過后,裸露金屬占測(cè)試面積的百分比,是評(píng)判涂層界面破壞類型并輔助判斷涂層與基底結(jié)合強(qiáng)度的重要參數(shù)[6,19]。從圖中可以發(fā)現(xiàn),在γ-APS/EP含量較低時(shí),剝離率隨附著力的增加而逐漸下降,在含量為3%時(shí)達(dá)到最小值3.27%,而含量過高反而使涂層剝離率升高。
圖3 涂層干態(tài)附著力和剝離率隨活性樹脂含量的變化
Fig.3 Dry adhesion strength and de-adhered ratio of coatings as a function of the content of γ-APS/EP
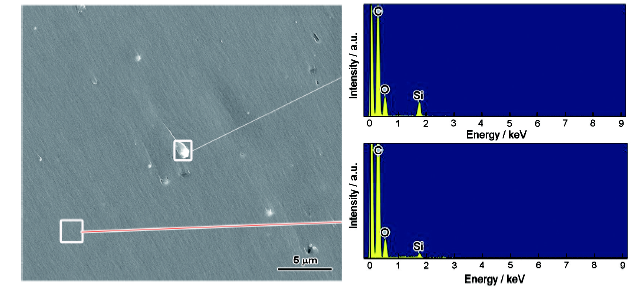
圖4所示為清漆和3%γ-APS/EP試樣干態(tài)附著力測(cè)試后金屬裸露區(qū)域背散射模式下的SEM像,黑色部分代表涂層,白色代表金屬基體,改性后金屬表面殘余的涂層覆蓋面積明顯增加。選擇金屬裸露區(qū)域有金屬光澤的區(qū)域進(jìn)行XPS測(cè)試,測(cè)試全譜和每種元素的分峰結(jié)果如圖5所示,每種元素峰的位置信息見表1[6,13,20]。從全譜可以發(fā)現(xiàn),添加了活性樹脂后,代表涂層信息的C1s、N1s和Si2p峰相對(duì)強(qiáng)度明顯增強(qiáng),證明附著力測(cè)試后金屬表面殘留了更多的涂層。分析XPS精細(xì)譜可以看到,Fe2O3的O1s峰相對(duì)強(qiáng)度顯著下降,同時(shí),在97~105 eV區(qū)間內(nèi),清漆金屬裸露區(qū)域光電子譜中沒有峰的跡象,而3%γ-APS/EP涂層金屬裸露區(qū)域在101.22和102.09 eV處出現(xiàn)了Si2p 1/2和Si2p 3/2峰。由于Fe2O3是金屬基底Q235表面的氧化物,且Si元素只能來自涂層中添加的活性樹脂,這說明添加了活性樹脂使得金屬表面殘存的涂層增多,進(jìn)一步證明了活性樹脂增強(qiáng)了涂層與金屬基底的附著力。
圖4 附著力測(cè)試后斷口金屬裸露區(qū)域背散射模式下的SEM像
Fig.4 SEM images in back scattered mode of pure epoxy (a) and 3%γ-APS/EP (b) after dry adhesion tests
圖5 清漆和3%γ-APS/EP涂層試樣附著力測(cè)試后金屬裸露處的XPS譜
Fig.5 XPS spectra of pure epoxy (a~c) and 3%γ-APS/EP (d~f) samples after dry adhesion test(a, d) survey (b, e) O1s (c, f) Si2p
表1 元素結(jié)合能和半峰寬
Table 1 XPS binding energy assignments and full width at half maximum (FWHM)
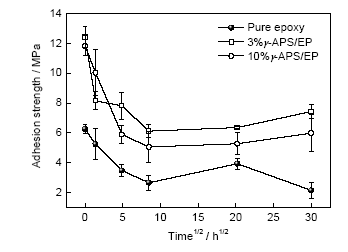
鑒于圖3干態(tài)附著力的變化規(guī)律,本工作對(duì)清漆、3%和10%活性樹脂3種典型涂層體系進(jìn)行進(jìn)一步研究。圖6為該3種涂層900 h內(nèi)濕態(tài)附著力隨時(shí)間的變化曲線。可以看出,涂層的濕態(tài)附著力在浸泡前期都會(huì)出現(xiàn)一個(gè)快速的下降,隨著浸泡時(shí)間的延長(zhǎng),涂層濕態(tài)附著力逐漸趨于穩(wěn)定。相較于清漆,添加了活性樹脂的涂層的濕態(tài)附著力在900 h內(nèi)都維持在一個(gè)較高的水平。結(jié)合浸泡900 h后涂層的拉拔斷口形貌(圖7)可以發(fā)現(xiàn),清漆試樣裸露出全部的金屬基體,為典型的界面破壞斷裂,而添加了3%和10%活性樹脂的試樣依舊有涂層覆蓋在測(cè)試區(qū)域,呈現(xiàn)出混合斷裂形式。說明清漆浸泡后涂層與金屬的結(jié)合變得很脆弱,基本喪失了對(duì)基底的保護(hù)作用,而加入活性樹脂能夠有效地減緩界面結(jié)合的劣化進(jìn)度,為金屬基底提供持續(xù)的防護(hù)。
圖6 清漆、3%γ-APS/EP和10%γ-APS/EP涂層的濕態(tài)附著力隨浸泡時(shí)間的變化
Fig.6 Wet adhesion strengths of pure epoxy, 3%γ-APS/EP and 10%γ-APS/EP coatings on Q235 panels as a function of immersion time in 3.5%NaCl solution
圖7 Q235鋼試樣經(jīng)900 h浸泡濕態(tài)附著力測(cè)試后的形貌
Fig.7 Surface morphologies of Q235 samples after 900 h wet adhesion strength test(a) pure epoxy (b) 3%γ-APS/EP (c) 10%γ-APS/EP
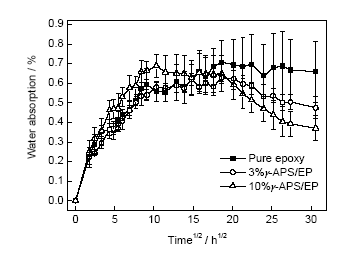
在保證涂層與金屬基體良好附著的前提下,涂層對(duì)腐蝕介質(zhì)優(yōu)異的抗?jié)B透性能可延緩附著力的損失[4]。涂層飽和吸水率測(cè)試是常用的抗?jié)B透性能測(cè)試方法,不同活性樹脂含量的環(huán)氧涂層吸水動(dòng)力學(xué)曲線如圖8所示。可以看出,環(huán)氧清漆的水傳輸動(dòng)力學(xué)明顯分為2個(gè)階段:快速吸收階段和飽和階段,而加入活性樹脂后,水傳輸增加了一個(gè)下降階段,典型時(shí)間點(diǎn)涂層的飽和吸水率數(shù)據(jù)如表2所示。在快速吸收階段,水通過涂層中的微觀通道擴(kuò)散迅速,吸水速率按由快到慢依次是:10%γ-APS/EP>3%γ-APS/EP>清漆。飽和吸水率由高到低依次是:清漆>10%γ-APS/EP>3%γ-APS/EP。由表2可以看出,吸水率到達(dá)飽和時(shí)間點(diǎn)的先后順序依次是:10%γ-APS/EP>3%γ-APS/EP>清漆。可見,清漆飽和吸水率高,證明自由體積分?jǐn)?shù)大或者界面缺陷多,能容納更大體積的水;而添加了3%活性樹脂后,涂層的吸水速率降低,同時(shí)涂層的飽和吸水率下降;但是當(dāng)活性樹脂含量增至10%時(shí),涂層吸水速率快且吸水量大,這可能是因?yàn)橐肓舜罅课捶磻?yīng)的甲氧基硅基團(tuán),能夠吸水并與之發(fā)生反應(yīng)。值得注意的是,3%γ-APS/EP和10%γ-APS/EP涂層的吸水率分別在406.5和346.5 h (表2)開始下降,可能是γ-APS/EP使涂層在浸泡后發(fā)生了結(jié)構(gòu)和成分上的改變[20],吸收的水又被排出體系。
圖8 涂層試樣的吸水動(dòng)力學(xué)曲線
Fig.8 Time dependence of water absorption for pure epoxy, 3%γ-APS/EP and 10%γ-APS/EP
表2 涂層在不同浸泡時(shí)間的吸水率
Table 2 Water absorptions of epoxy coatings with different immersion time
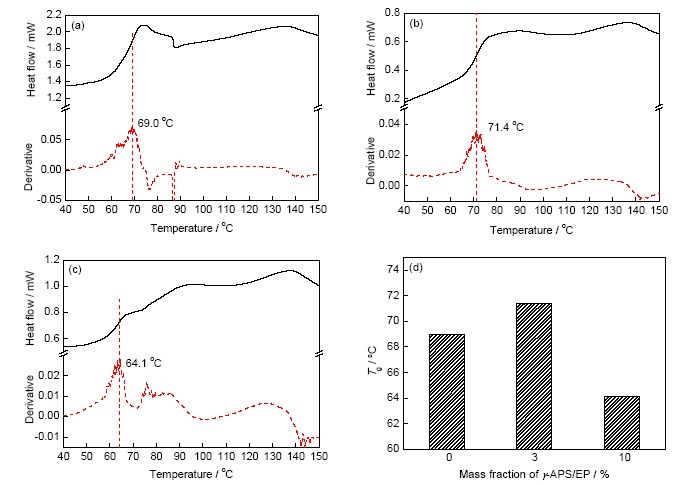
在涂層完整無缺陷(裂紋和小孔)的情況下,H2O和O2可通過涂層內(nèi)的自由體積進(jìn)行傳輸。在涂層體系確定的情況下(環(huán)氧胺體系),自由體積百分比取決于涂層的交聯(lián)密度,高交聯(lián)密度意味著小尺寸的自由體積和小的自由體積分?jǐn)?shù),有助于提高抗?jié)B性能。玻璃化轉(zhuǎn)變溫度(Tg)是判斷高聚物交聯(lián)程度的重要參數(shù):交聯(lián)密度越高,自由體積變小因而分子鏈段趨于凍結(jié),材料的Tg越高[21]。圖9為涂層浸泡前的DSC熱流曲線及相應(yīng)的一階微分曲線,將對(duì)應(yīng)微分曲線的最大值處的溫度作為涂層的Tg。可以看到,加入3%的活性樹脂提升了涂層的Tg,由清漆的69.0 ℃上升達(dá)到71.4 ℃,即涂層的交聯(lián)密度略有升高,但活性樹脂的含量為10%時(shí),Tg大幅下降,僅為64.1 ℃,說明活性樹脂含量過多減小了涂層的交聯(lián)密度。
圖9 涂層DSC熱流-溫度曲線及玻璃化轉(zhuǎn)變溫度(Tg)
Fig.9 DSC curves of pure epoxy (a), 3%γ-APS/EP (b), 10%γ-APS/EP (c) and corresponding glass transition temperature (Tg) (d)
圖10所示為玻璃樣品在背散射模式下的縱截面SEM像。加入活性樹脂后,涂層內(nèi)部出現(xiàn)了一些白色點(diǎn)(圖10c和e),并隨著γ-APS/EP含量的上升而增加,在浸泡之后數(shù)量呈上升趨勢(shì)(圖10d和f)。圖11為白點(diǎn)區(qū)域和涂層其它區(qū)域的EDS分析。對(duì)比發(fā)現(xiàn),白點(diǎn)區(qū)域和其它區(qū)域的Si和O元素質(zhì)量比分別為0.28和0.07;同時(shí)從元素面分析圖(圖12)也可觀察到白點(diǎn)處的Si元素和O元素襯度高于其它區(qū)域,均說明白點(diǎn)區(qū)域是Si和O 2種元素的富集區(qū)。由于這2種元素只可能來自γ-APS/EP,所以白點(diǎn)應(yīng)該是硅羥基縮聚生成的聚硅氧烷。浸泡后白點(diǎn)數(shù)量增加,這可能是由于浸泡引起一些甲氧基硅官能團(tuán)進(jìn)一步水解并發(fā)生縮合反應(yīng)造成的。
圖10 清漆、3%γ-APS/EP和10%γ-APS/EP涂層浸泡900 h前后縱截面SEM像
Fig.10 Cross-sectional SEM images of pure epoxy (a, b), 3%γ-APS/EP (c, d) and 10%γ-APS/EP (e, f) coatings before (a, c, e) and after (b, d, f) 900 h immersion in 3.5%NaCl solution
圖11 10%γ-APS/EP涂層縱截面EDS結(jié)果
Fig.11 SEM-EDS analysis of 10%γ-APS/EP coating
圖12 10% g-APS/EP涂層縱截面形貌及對(duì)應(yīng)EDS面分析結(jié)果
Fig.12 Cross-sectional morphology of 10% g-APS/EP and the corresponding EDS mappings
聚合物內(nèi)部引入的一些小分子會(huì)對(duì)聚合物起到塑化的作用,表現(xiàn)為Tg的下降。涂層中加入活性樹脂對(duì)涂層結(jié)構(gòu)的影響是復(fù)雜的。一方面,水解后的硅羥基既可以與樹脂分子鏈上的羥基進(jìn)行反應(yīng),又可自行縮聚,同時(shí)活性樹脂上的環(huán)氧基與固化劑的氨基具有反應(yīng)活性,這都有利于涂層交聯(lián)密度的提升,使得涂層Tg提高。另一方面,當(dāng)活性樹脂含量增加時(shí),硅羥基數(shù)量增加導(dǎo)致縮聚生成聚硅氧烷第二相(白點(diǎn)),會(huì)隔開交聯(lián)體系單元分子鏈,隔離作用導(dǎo)致涂層塑化。在γ-APS/EP含量等于3%時(shí),交聯(lián)密度提升占主導(dǎo),所以Tg升高;γ-APS/EP含量為10%時(shí),塑化作用凸顯,導(dǎo)致Tg下降。
綜上可知,當(dāng)γ-APS/EP添加量為3%時(shí),環(huán)氧樹脂涂層與碳鋼基體有最好的附著性能,且涂層表現(xiàn)出對(duì)NaCl腐蝕介質(zhì)良好的抗?jié)B性能。為了分析其中的機(jī)理,需要了解涂料制備和成膜過程中一切可能發(fā)生的反應(yīng)。在涂料制備階段,由于涂料在高速攪拌過程中會(huì)卷入帶有一定濕度的空氣,使活性樹脂發(fā)生部分水解[22,23],如圖13的反應(yīng)1所示。由于硅羥基具有很高的反應(yīng)活性,它自身能進(jìn)一步縮聚生成聚硅氧烷[22,23,24](圖13中反應(yīng)2)。所以,當(dāng)活性樹脂含量增加,硅羥基濃度上升,會(huì)加快反應(yīng)速率并導(dǎo)致反應(yīng)產(chǎn)物聚硅氧烷的量上升(圖10中白點(diǎn)數(shù)量的增多)。在涂刷成膜階段,加入固化劑以后,一方面活性樹脂有機(jī)端環(huán)氧基會(huì)與固化劑發(fā)生反應(yīng)(圖13中反應(yīng)3),增強(qiáng)了活性樹脂與環(huán)氧涂層的相容性,另一方面涂料制備階段未反應(yīng)的硅羥基會(huì)通過氫鍵作用先吸附在金屬表面,后期脫水后與金屬表面的羥基反應(yīng)生成噁烷鍵[25,26],如圖13中反應(yīng)4所示,實(shí)現(xiàn)了環(huán)氧涂層與金屬界面的化學(xué)鍵合。同時(shí),吸附在金屬表面相鄰的活性樹脂還會(huì)發(fā)生縮聚,形成一層疏水的結(jié)構(gòu)[11,26]。據(jù)此,活性樹脂在金屬表面的吸附情況可分為以下3種:
(1) 亞飽和吸附狀態(tài)。活性樹脂含量較少,在3%以下時(shí)(如0.5%和1%),活性樹脂濃度低,體系中的水含量雖然足以使得活性樹脂的水解反應(yīng)較為完全(圖13反應(yīng)1),但水解后的活性樹脂受總含量的限制,加上自身縮聚成聚硅氧烷的影響,在金屬表面化學(xué)鍵合的數(shù)量較少,呈亞飽和吸附狀態(tài),使得涂層的附著力較環(huán)氧清漆有所提高(圖3),但并未達(dá)到峰值。
(2) 飽和吸附狀態(tài)。活性樹脂含量為3%時(shí),活性樹脂在發(fā)生圖13反應(yīng)1中的水解反應(yīng)后,雖然自聚縮合生成了一定量的聚硅氧烷結(jié)構(gòu),但仍有足夠數(shù)量水解后的活性樹脂與金屬表面的羥基發(fā)生化學(xué)鍵合,此時(shí)活性樹脂在金屬界面呈飽和吸附狀態(tài),環(huán)氧涂層與碳鋼的附著力達(dá)到峰值。
(3) 過飽和水解狀態(tài)。活性樹脂含量大于3%時(shí)(如5%和10%),活性樹脂濃度過高,大量的活性樹脂不能發(fā)生水解。發(fā)生水解的活性樹脂縮聚程度達(dá)到最高,聚硅氧烷的數(shù)量達(dá)到峰值,造成能夠與金屬表面進(jìn)行物理吸附的硅羥基數(shù)量減少,導(dǎo)致環(huán)氧涂層與金屬間的化學(xué)鍵合數(shù)量減少,附著力下降(圖3)。
在環(huán)氧涂層服役的過程中,未水解的甲氧基硅官能團(tuán)數(shù)量隨著活性樹脂含量的增加總體呈上升趨勢(shì),這類基團(tuán)對(duì)涂層的抗?jié)B性能有很大的影響。在涂層吸水后,甲氧基硅官能團(tuán)能夠發(fā)生水解反應(yīng)(圖13反應(yīng)1),并相繼發(fā)生縮聚反應(yīng)(圖13反應(yīng)2)使涂層的致密度提高。水解反應(yīng)加快涂層的前期吸水速率,縮聚反應(yīng)導(dǎo)致后期涂層的吸水率下降,這也就解釋了圖8中飽和吸水率前期快速上升和后期下降的原因,同時(shí)說明了環(huán)氧涂層經(jīng)浸泡后聚硅氧烷數(shù)量增加的緣由(圖10)。
圖13 活性樹脂的作用機(jī)理示意圖
Fig.13 Mechanism of γ-APS/EP action
(1) 利用γ-APS的氨基與環(huán)氧樹脂的環(huán)氧基開環(huán)反應(yīng),成功制備了一種具有水解特性的活性樹脂,活性樹脂加入環(huán)氧涂層中能夠提升環(huán)氧樹脂與金屬基體的界面結(jié)合力,同時(shí)增加樹脂對(duì)外界腐蝕介質(zhì)的抗?jié)B性能。
(2) 活性樹脂在涂層中存在自縮聚和化學(xué)鍵合2種競(jìng)爭(zhēng)過程,活性樹脂含量為3%時(shí),環(huán)氧涂層與金屬基體界面的結(jié)合力最佳,為環(huán)氧清漆樹脂的2倍,活性樹脂含量過少或過多均會(huì)減少環(huán)氧涂層/碳鋼表面化學(xué)鍵合的數(shù)量。
(3) 隨活性樹脂含量的升高,涂層對(duì)外界腐蝕介質(zhì)的抗?jié)B透性能先下降后升高,活性樹脂含量為3%時(shí)涂層的交聯(lián)密度最高,飽和吸水率最低。活性樹脂含量過高,聚硅氧烷異質(zhì)相塑化涂層使涂層最大吸水率升高,且涂層內(nèi)部剩余過量的甲氧基硅基團(tuán)易水解,會(huì)快速地吸水使涂層達(dá)到飽和。
 , 楊延格, 曹京宜
, 楊延格, 曹京宜
1 實(shí)驗(yàn)方法
1.1 實(shí)驗(yàn)材料
1.2 活性樹脂的合成
1.3 試樣制備
1.4 涂層吸水率測(cè)試
1.5 附著力測(cè)試
1.6 結(jié)構(gòu)與成分表征
2 實(shí)驗(yàn)結(jié)果與分析
2.1 活性樹脂γ-APS/EP的合成機(jī)理
2.2 活性樹脂對(duì)涂層/碳鋼界面結(jié)合力的影響


Peak
Bond
Pure epoxy
3%γ-APS/EP
Binding energy
FWHM
Binding energy
FWHM
O1s
Si—O/C—O[6,13]
529.772
1.051
529.825
1.237
—OH[6]
531.285
1.557
531.369
1.644
Fe2O3[20]
532.728
1.836
532.855
1.544
Si2p 1/2
Si—O[13]
101.220
2.063
Si2p 3/2
102.090
2.192

2.3 活性樹脂對(duì)涂層抗?jié)B透性能的影響
Mass fraction of
γ-APS/EP / %
Water absorption / %
Saturated time / h
Decline time / h
11 h
70.08 h
106 h
346 h
936 h
0
0.3537
0.5715
0.5567
0.7067
0.6582
309
-
3
0.2925
0.5328
0.5788
0.6266
0.4737
106
406.5
10
0.3553
0.6587
0.6888
0.6479
0.3701
70.08
346.5


2.4 活性樹脂對(duì)環(huán)氧涂層/碳鋼界面的化學(xué)鍵合機(jī)理分析

3 結(jié)論
來源--金屬學(xué)報(bào)
“推薦閱讀”
【責(zé)任編輯】:國(guó)檢檢測(cè)版權(quán)所有:轉(zhuǎn)載請(qǐng)注明出處
 滬公網(wǎng)安備31011202020290號(hào)
滬公網(wǎng)安備31011202020290號(hào)
